
Abstract
Over the past two decades, the molecular machinery that underlies autophagic responses has been characterized with ever increasing precision in multiple model organisms. Moreover, it has become clear that autophagy and autophagy‐related processes have profound implications for human pathophysiology. However, considerable confusion persists about the use of appropriate terms to indicate specific types of autophagy and some components of the autophagy machinery, which may have detrimental effects on the expansion of the field. Driven by the overt recognition of such a potential obstacle, a panel of leading experts in the field attempts here to define several autophagy‐related terms based on specific biochemical features. The ultimate objective of this collaborative exchange is to formulate recommendations that facilitate the dissemination of knowledge within and outside the field of autophagy research.
Similar content being viewed by others

Introduction
The Nobel Assembly at Karolinska Institute awarded the 2016 Prize in Physiology or Medicine to the cell biologist Yoshinori Ohsumi for his early identification and characterization of the autophagy machinery, in particular, AuTophaGy‐related (Atg) genes, in yeast (Tsukada & Ohsumi, 1993). This came as an overt recognition to a field symbolically initiated by the Belgian cytologist and biochemist Christian De Duve, who in 1963 employed the term autophagy (from the Ancient Greek αὐτόφαγος, meaning “self‐eating”) for describing the presence of single‐ or double‐membraned intracellular vesicles that contain parts of the cytoplasm and organelles in various states of disintegration (Yang & Klionsky, 2010). Our understanding of autophagy, which is highly conserved during evolution (Table 1), has tremendously expanded over the past decades, on both mechanistic and pathophysiological grounds (Choi et al, 2013; Noda & Inagaki, 2015). In parallel, we have begun to appreciate the considerable potential of pharmacological agents or dietary interventions that activate or inhibit autophagy as novel therapies for multiple human disorders and pathophysiological conditions, including neurodegenerative (Menzies et al, 2015), infectious (Deretic et al, 2013), autoimmune (Deretic et al, 2013; Zhong et al, 2016), cardiovascular (Shirakabe et al, 2016), rheumatic (Rockel & Kapoor, 2016), metabolic (Kim & Lee, 2014), pulmonary (Nakahira et al, 2016), and malignant diseases (Galluzzi et al, 2015b, 2017a; Amaravadi et al, 2016), as well as aging (Melendez et al, 2003; Lapierre et al, 2015; Lopez‐Otin et al, 2016). Nevertheless, there is not a single drug currently licensed by the US Food and Drug Administration (FDA)—or equivalent regulatory agency—that was developed with the primary aim of modulating autophagy (although many FDA‐approved drugs indeed activate or inhibit autophagy to some extent) (Poklepovic & Gewirtz, 2014; Rosenfeld et al, 2014; Vakifahmetoglu‐Norberg et al, 2015). Such a barrier in the translation of robust preclinical data from multiple model organisms into clinically viable therapeutic interventions reflects the persistence of several obstacles of pharmacological, biological, and technological nature. Discussing these issues in a comprehensive manner goes well beyond the scope of the current article and has been done elsewhere (Galluzzi et al, 2017b). An analysis of the literature also reveals considerable confusion about the use of several autophagy‐related terms, affecting not only less‐experienced investigators but also researchers with many years of experience in the field. Although such a semantic issue may appear trivial at first glance, we are concerned that it may constitute a significant obstacle to the optimal development of autophagy research, both at preclinical and translational levels. This problem has been overtly recognized and discussed throughout the past year. Starting from such a constructive exchange and driven by the success obtained by a similar initiative in the cell death field (Galluzzi et al, 2012, 2015a), leading experts in autophagy decided to gather and tentatively define several autophagy‐related terms based on precise biochemical features of the process.
Processes
Autophagy
Perhaps surprisingly, the relatively broad term “autophagy” itself has been used with rather variable and sometimes misleading connotations. We agree on two main features that characterize bona fide, functional autophagic responses, irrespective of type: (i) they involve cytoplasmic material; and (ii) they culminate with (and strictly depend on) lysosomal degradation. Thus, although autophagy substrates (see below for a definition) can be endogenous, such as damaged mitochondria and nuclear fragments, or exogenous, such as viruses or bacteria escaping phagosomes, autophagy operates on entities that are freely accessible to cytosolic proteins (notably, components of the autophagy machinery). This feature is important in order to discriminate autophagic responses from branches of vesicular trafficking that originate at the plasma membrane, which also culminates in lysosomal degradation. Such endocytic processes (which have cumulatively been referred to as “heterophagy” in the past) include phagocytosis (i.e., the uptake of particulate material by professional phagocytes—such as macrophages and immature dendritic cells—or other cells), receptor‐mediated endocytosis (i.e., the uptake of extracellular material driven by plasma membrane receptors), and pinocytosis (i.e., the relatively non‐specific uptake of extracellular fluids and small molecules) (Munz, 2017; Foot et al, 2017). However, some forms of autophagy (notably macroautophagy and endosomal microautophagy, see below for definitions) and the endocytic pathway interact at multiple levels, and the molecular machinery responsible for the fusion of late endosomes (also known as multi‐vesicular bodies) or autophagosomes (see below for a definition) with lysosomes is essentially the same (Tooze et al, 2014).
The strict dependency of autophagic responses on lysosomal activity is important to discriminate them from other catabolic pathways that also involve cytoplasmic material, such as proteasomal degradation (Bhattacharyya et al, 2014). The 26S proteasome degrades a large number of misfolded cytoplasmic proteins that have been ubiquitinated, as well as properly folded proteins that expose specific degradation signals, such as the so‐called N‐degrons (Sriram et al, 2011). When ubiquitinated proteins accumulate, however, they tend to assemble into aggregates that are degraded by macroautophagy upon binding to autophagy receptors (see below for a definition) (Lim & Yue, 2015; Moscat et al, 2016). Moreover, considerable cross talk between the proteasome and chaperone‐mediated autophagy (CMA, see below for a definition) has been described (Massey et al, 2006; Schneider et al, 2014), and cytosolic proteins bound to heat shock protein family A (Hsp70) member 8 (HSPA8), which serves as the main chaperone in CMA, can be efficiently redirected to proteasomal degradation upon interaction with ubiquilin 2 (UBQLN2) (Hjerpe et al, 2016). Thus, the proteasome system shares some substrates with different forms of autophagy. However, these two catabolic pathways differ radically in their final products. Proteasomal degradation results in short peptides (of 8–12 residues) that are not necessarily degraded further, but may feed into additional processes including (but not limited to) antigen presentation at the plasma membrane (Neefjes et al, 2011). In contrast, lysosomal proteases fully catabolize polypeptides to their constituting amino acids, which eventually become available for metabolic reactions or repair processes. Moreover, lysosomal hydrolases also degrade lipids, sugars, and nucleic acids (Settembre et al, 2013). In summary, bona fide functional autophagic responses direct cytoplasmic material of endogenous or exogenous origin to degradation within lysosomes (or late endosomes, in specific cases).
Microautophagy and endosomal microautophagy
Microautophagy is a form of autophagy during which cytoplasmic entities destined for degradation are directly taken up by the vacuole (in yeast and plants) via direct membrane invagination (Farre & Subramani, 2004; Uttenweiler & Mayer, 2008). In cells from Drosophila melanogaster and mammals, a similar mechanism involves late endosomes. This process, which also occurs in yeast cells, is commonly known as “endosomal microautophagy” (Sahu et al, 2011; Uytterhoeven et al, 2015; Mukherjee et al, 2016). In yeast, microautophagy has been involved in the degradation of multiple substrates, including peroxisomes (a process called “micropexophagy”, historically the first form of yeast microautophagy to be described) (Farre & Subramani, 2004), portions of the nucleus (Kvam & Goldfarb, 2007), damaged mitochondria (Kissova et al, 2007), and lipid droplets (Vevea et al, 2015). In plants, microautophagy has been shown to mediate the degradation of anthocyanins (Chanoca et al, 2015). Finally, endosomal microautophagy degrades cytosolic proteins, either in bulk or selectively (only proteins containing a KFERQ‐like motif recognized by HSPA8) (Sahu et al, 2011; Uytterhoeven et al, 2015; Mukherjee et al, 2016). Of note, some proteins internalized by multivesicular bodies through direct membrane invagination can be spared from degradation and released in the extracellular microenvironment within exosomes (Record et al, 2014).
Arguably, microautophagy is the least studied form of autophagy, but a molecular signature of the process has begun to emerge. Thus, several forms of yeast microautophagy (e.g., micropexophagy) require some components of the macroautophagy machinery for cargo targeting and internalization, including (but perhaps not limited to) Atg7, Atg8, and Atg9 (Farre et al, 2008; Krick et al, 2008). Conversely, endosomal microautophagy relies on multiple endosomal sorting complexes required for transport (ESCRT) systems (Sahu et al, 2011; Liu et al, 2015b; Uytterhoeven et al, 2015; Mukherjee et al, 2016). In addition, the selective uptake of KFERQ‐containing proteins by late endosomes in the course of endosomal microautophagy depends on HSPA8, reflecting its ability to directly interact with phosphatidylserine on (and hence deform) the outer endosomal membrane (Uytterhoeven et al, 2015; Morozova et al, 2016). Along similar lines, chaperone ATPase HSP104 (Hsp104) reportedly underlies microautophagic responses to lipid droplets in Saccharomyces cerevisiae (Vevea et al, 2015). However, the strict requirement of chaperones from the HSP70 protein family in other variants of microautophagy has not yet been documented. Of note, the yeast orthologue of mammalian NBR1, autophagy cargo receptor (NBR1; which is known to operate as a macroautophagy receptor, see below) reportedly underlies an ESCRT‐dependent and ubiquitination‐dependent microautophagic pathway in Schizosaccharomyces pombe (Liu et al, 2015b). It will be interesting to determine whether NBR1 and other components of this pathway also contribute to microautophagy in mammalian cells. Irrespectively, we propose to define microautophagy and endosomal microautophagy as types of autophagy in which the cargo is directly internalized in small vesicles that form at the surface of the lysosome/vacuole or late endosomes (multivesicular bodies), respectively, via ESCRT‐independent (microautophagy) or ESCRT‐dependent (endosomal microautophagy), mechanisms. In addition, selective endosomal microautophagy can be defined as an HSPA8‐dependent autophagic response, but it can be differentiated from CMA based on (i) its dependence on ESCRT systems and (ii) its independence from a specific splicing variant of lysosomal‐associated membrane protein 2 (LAMP2A, see below) (Table 1).
Chaperone‐mediated autophagy
CMA involves the direct delivery of cytosolic proteins targeted for degradation to the lysosome (Kaushik & Cuervo, 2012). The distinctive feature of CMA is that neither vesicles nor membrane invaginations are required for substrate delivery to lysosomes, since substrates reach the lysosomal lumen through a protein‐translocation complex at the lysosomal membrane (Kaushik & Cuervo, 2012). CMA only degrades soluble proteins bearing a KFERQ‐like motif bound to HSPA8 (Dice, 1990), but not organelles, other macromolecules such as lipids, nucleic acids, or proteins integral to membranes (Chiang et al, 1989; Wing et al, 1991; Salvador et al, 2000). CMA has been shown to operate on a multitude of cytosolic proteins, hence exerting major regulatory functions in different pathophysiological scenarios such as metabolic regulation (Schneider et al, 2014; Kaushik & Cuervo, 2015), genome integrity preservation (Park et al, 2015), aging (Cuervo & Dice, 2000; Rodriguez‐Muela et al, 2013; Schneider et al, 2015), T‐cell activation (Valdor et al, 2014), neurodegeneration (Orenstein et al, 2013), and oncogenesis (Kon et al, 2011). Moreover, linear sequence analysis of the cytosolic proteome suggests that ~30% of its components may be degraded by CMA (Dice, 1990). Importantly, the translocation of CMA substrates across the lysosomal membrane relies on a dedicated molecular machinery that critically involves a specific splicing isoform of LAMP2, namely, LAMP2A (Cuervo & Dice, 1996). Thus, chaperone‐bound autophagy substrates bind LAMP2A monomers on the cytosolic side of the lysosome, which stimulate the formation of an oligomeric LAMP2A translocation complex (Bandyopadhyay et al, 2008).
While unfolding and dissociating from chaperones (Salvador et al, 2000), CMA substrates are translocated into the lysosomal lumen through oligomeric LAMP2A complexes that are stabilized by a lysosomal pool of heat shock protein 90 alpha family class A member 1 (HSP90AA1; best known as HSP90) (Bandyopadhyay et al, 2008), and a cytosolic pool of glial fibrillary acidic protein (GFAP) (Bandyopadhyay et al, 2010). Lysosomal HSPA8 operates as an acceptor for CMA substrates, possibly by preventing cytosolic retrotranslocation (Agarraberes et al, 1997). Eventually, LAMP2A complexes are dismantled within lipid‐rich microdomains of the lysosomal membrane by a mechanism that relies on HSPA8, followed by cathepsin A (CTSA)‐catalyzed LAMP2A degradation (Kaushik et al, 2006). The CMA‐supporting activity of GFAP is negatively regulated by phosphorylation, which is catalyzed by a pool of AKT serine/threonine kinase 1 (AKT1) that resides on the lysosomal surface (Arias et al, 2015). In this setting, dephosphorylation of AKT1 by PH domain and leucine‐rich repeat protein phosphatase 1 (PHLPP1) counteracts the tonic activity of mechanistic target of rapamycin (MTOR) complex 2 (mTORC2), resulting in CMA activation (Arias et al, 2015). It remains to be determined to what extent CMA is conserved in lower organisms, since the splice variant of LAMP2 that is essential for CMA (i.e., LAMP2A) appeared relatively late in evolution (i.e., in birds) (Eskelinen et al, 2005). It has been suggested that selective endosomal microautophagy, which shares with CMA the dependence on KFERQ‐like motives and HSPA8, constitutes an alternative to CMA in D. melanogaster (Mukherjee et al, 2016). Irrespective of this unknown, we propose to define CMA as an HSPA8‐dependent autophagic response that relies on LAMP2A‐mediated cargo translocation across the lysosomal membrane. In this context, it should be noted that other splicing isoforms of LAMP2 (including LAMP2B and LAMP2C) are dispensable for CMA but involved in macroautophagy (see below) (Eskelinen et al, 2005). This implies that genetic interventions aimed at specifically inhibiting CMA should not be directed to HSPA8 (which is also required for multiple forms of microautophagy), nor to LAMP2 as a gene (Table 1).
Macroautophagy
Macroautophagy is the variant of autophagy best characterized thus far, at least in part owing to its easily distinguishable morphological features. Indeed, whereas microautophagy and CMA are not associated with major morphological changes in vesicular compartments, macroautophagic responses involve dedicated vesicles that can occupy (at a specific moment) a considerable part of the cytoplasm, an impressive phenomenon that attracted attention as early as in the late 1950s (Yang & Klionsky, 2010). These double‐membraned vesicles, which are commonly known as autophagosomes, can sequester large portions of the cytoplasm including entire organelles or parts thereof. This endows macroautophagy with a considerable catabolic potential that—in specific settings—can contribute to regulated cell death (RCD) (Galluzzi et al, 2016) or cellular atrophy leading to neurodegeneration (Cherra et al, 2010a,b; Zhu et al, 2013). The molecular machinery that executes and regulates macroautophagy in organisms encompassing yeast, nematodes, flies, and mammals has been the subject of intense investigation throughout the past two decades (Noda & Inagaki, 2015; Antonioli et al, 2016). Although a detailed description of these pathways is not warranted here, a few functional modules of the macroautophagy apparatus are particularly important for this discussion. Indeed, the molecules that are part of these functional modules, their interactors and the processes they control have been extensively employed thus far to identify macroautophagic responses, though not always with precision. Efficient macroautophagic responses involving the formation of autophagosomes, their fusion with lysosomes, and lysosomal degradation have been associated with the activity of two ubiquitin‐like conjugation systems (Noda & Inagaki, 2015; Antonioli et al, 2016). One relies on ATG7 and ATG10, which promote the conjugation of ATG5 to ATG12 in the context of a multiprotein complex containing autophagy‐related 16‐like 1 (ATG16L1) (Mizushima et al, 1998). Another one is mediated by ATG3 and ATG7, which together with the ATG12‐ATG5:ATG16L1 complex conjugates phosphatidylethanolamine to microtubule‐associated protein 1 light chain 3 beta (MAP1LC3B; best known as LC3B) and other orthologues of yeast Atg8 upon ATG4‐dependent proteolytic maturation (Ichimura et al, 2000; Marino et al, 2010; Rockenfeller et al, 2015). Lipidated LC3 (often referred to as LC3‐II) is generated onto forming autophagosomes and allows for substrate uptake upon binding to several autophagy receptors (Kabeya et al, 2000; Stolz et al, 2014; Wild et al, 2014). Importantly, robust data suggest that the ATG conjugation systems and Atg8‐like proteins are not strictly required for the formation of autophagosomes, as classically thought (although their absence greatly reduces the efficiency of the process), but also contribute to autophagosome extension around large substrates and closure, the fusion of autophagosomes with lysosomes, and the degradation of the inner autophagosomal membrane (Nguyen et al, 2016; Tsuboyama et al, 2016).
In response to commonly studied stimuli including starvation, autophagosome formation is initiated by the assembly and activation of a multiprotein complex containing ATG13, ATG101, RB1 inducible coiled‐coil 1 (RB1CC1; best known as FIP200) and unc‐51‐like autophagy activating kinase 1 (ULK1, the mammalian orthologue of yeast Atg1) at ATG9‐containing membranes, followed by ULK1‐dependent ATG9 phosphorylation (Orsi et al, 2012; Papinski et al, 2014; Stanley et al, 2014; Joachim et al, 2015; Karanasios et al, 2016). This event initiates the elongation of pre‐autophagosomal membranes upon incorporation of phospholipids from various sources including the endoplasmic reticulum (ER), recycling endosomes, and mitochondria (Lamb et al, 2013), and allows for the recruitment of a multiprotein complex with Class III phosphatidylinositol 3‐kinase (PI3K) activity, which contains beclin 1 (BECN1), phosphatidylinositol 3‐kinase catalytic subunit type 3 (PIK3C3; best known as VPS34), phosphoinositide‐3‐kinase regulatory subunit 4 (PI3KR4; best known as VPS15) (Kihara et al, 2001a,b), the sensor of membrane curvature ATG14 (also known as ATG14L or BARKOR) (Itakura et al, 2008; Sun et al, 2008; Matsunaga et al, 2009; Zhong et al, 2009; Fan et al, 2011), and nuclear receptor binding factor 2 (NRBF2) (Lu et al, 2014a). On activation, VPS34 produces phosphatidylinositol 3‐phosphate (PI3P), which further supports the expansion of autophagosomal membranes until closure by engaging PI3P‐binding ATG proteins and members of the WIPI family (Proikas‐Cezanne et al, 2015). Both the ULK1 and autophagy‐specific Class III PI3K complexes are highly regulated. One of the main regulators of macroautophagy is MTOR complex 1 (mTORC1), which robustly suppresses autophagosome formation by catalyzing the inactivating phosphorylation of ATG13 and ULK1 (Jung et al, 2009; Nicklin et al, 2009; Nazio et al, 2013) Moreover, mTORC1 inhibits macroautophagic responses by preventing the nuclear translocation of transcription factor EB (TFEB, a master transcriptional regulator of lysosomal biogenesis and macroautophagy) upon phosphorylation on S142 (Settembre et al, 2011, 2012). Such a multipronged inhibitory network is disrupted upon mTORC1 inactivation by AMP‐activated protein kinase (AMPK), which responds to reduced ATP levels and consequent AMP accumulation (Inoki et al, 2002). AMPK also catalyzes activating phosphorylation events on ULK1 (Lee et al, 2010; Egan et al, 2011; Kim et al, 2011) and BECN1 (Kim et al, 2013b). In mammalian cells, ULK1 directly phosphorylates BECN1, resembling AMPK in its VPS34‐stimulatory effects (Russell et al, 2013), and ATG14 (Park et al, 2016; Wold et al, 2016). The autophagy‐specific Class III PI3K complex is regulated by several interactors, including the VPS34 activator autophagy and beclin 1 regulator 1 (AMBRA1, originally “activating molecule in Beclin 1‐regulated autophagy”), as well as the BECN1 inhibitor BCL2, which also interacts with ATG12 (Liang et al, 1999; Pattingre et al, 2005; Fimia et al, 2007; Zalckvar et al, 2009; Rubinstein et al, 2011).
Once autophagosomes have enclosed autophagy substrates, they can fuse with late endosomes or lysosomes to form amphisomes or autolysosomes (see below for definitions). The molecular machinery that is responsible for these fusion events involve dozens of proteins, most of which are shared with the endocytic pathway (Amaya et al, 2015; Antonioli et al, 2016). In this setting, an important role is mediated by the activation of the GTPase RAB7A, member RAS oncogene family (RAB7A), which is required for autophagosome maturation (Gutierrez et al, 2004; Jager et al, 2004; Liang et al, 2008), the RAB7 effector pleckstrin homology and RUN domain containing M1 (PLEKHM1) (McEwan et al, 2015), the PI3P‐binding protein tectonin beta‐propeller repeat containing 1 (TECPR1) (Chen et al, 2012), ectopic P‐granules autophagy protein 5 homolog (EPG5) (Tian et al, 2010), inositol polyphosphate‐5‐phosphatase E (INPP5E) (Hasegawa et al, 2016), syntaxin 17 (STX17), and other soluble N‐ethylmaleimide‐sensitive factor activating protein receptor (SNARE) proteins (Fader et al, 2009; Nair et al, 2011; Itakura et al, 2012), as well as homotypic fusion and vacuole protein sorting (HOPS) complexes (McEwan et al, 2015). ATG14, LAMP2B (but not LAMP2A) as well as phosphorylated and lipidated LC3 are also involved in the formation of autolysosomes (Eskelinen et al, 2005; Diao et al, 2015; Wilkinson et al, 2015; Nguyen et al, 2016). Conversely, RUN and cysteine‐rich domain containing beclin 1 interacting protein (RUBCN; best known as RUBICON) negatively regulates the fusion of autophagosomes with lysosomes upon interacting with VPS34 (Matsunaga et al, 2009). Degradation of autophagy substrates proceeds as the lysosomal lumen is acidified (owing to the activity of an ATP‐dependent proton pump commonly known as V‐type ATPase) (Mindell, 2012), upon disassembly of the inner autophagosomal membrane supported by the ATG conjugation systems (Tsuboyama et al, 2016). Finally, mTORC1 reactivation inhibits macroautophagy as it promotes so‐called autophagic lysosome reformation (ALR), a process whereby proto‐lysosomal vesicles extruding from autolysosomes mature to regenerate the lysosomal compartment (Yu et al, 2010).
Several of the proteins mentioned above including ATG3, ATG5, ATG7, ATG9, ATG13, ATG16L1, ULK1, BECN1, and VPS34 have been considered as strictly required for macroautophagic responses (irrespective of their functions in autophagy‐independent processes) (Codogno et al, 2012). At least in part, such a view originated from the embryonic or post‐natal lethality caused in mice by the genetic ablation of any of these components of the macroautophagy machinery at the whole‐body level (Qu et al, 2003; Yue et al, 2003; Kuma et al, 2004; Komatsu et al, 2005; Gan et al, 2006; Saitoh et al, 2008, 2009; Sou et al, 2008), which is likely to reflect the key role of macroautophagy in development and adult tissue homeostasis (although such a general phenotype might also stem from autophagy‐independent functions of these proteins). In addition, both pharmacological and genetic interventions targeting these and other components of the macroautophagy apparatus have been associated with autophagic defects in hundreds of experimental settings, in vitro and in vivo. However, the discovery of bona fide macroautophagic responses occurring independently of ATG3, ATG5, ATG7, ULK1, BECN1, VPS34, and its product (PI3P) (Zhu et al, 2007; Nishida et al, 2009; Chang et al, 2013; Niso‐Santano et al, 2015; Vicinanza et al, 2015) casted doubts on the exclusive requirement of these factors for all forms of macroautophagy (Klionsky et al, 2016). The existence of ATG3‐, ATG5‐, ATG7‐, ULK1‐, BECN1‐, VPS34‐, and PI3P‐independent forms of macroautophagy lent further support to the hypothesis that the molecular mechanisms underlying macroautophagic responses exhibit considerable degree of redundancy (at least in mammals) (Nishida et al, 2009; Chu, 2011; Chang et al, 2013; Niso‐Santano et al, 2015; Vicinanza et al, 2015). This notion had previously been postulated based on the observation that some components of the macroautophagy apparatus have multiple functional homologues. For instance, the human genome codes for at least six distinct Atg8‐like proteins, namely microtubule‐associated protein 1 light chain 3 alpha (MAP1LC3A; best known as LC3A), LC3B, microtubule‐associated protein 1 light chain 3 gamma (MAP1LC3C; best known as LC3C), GABA type A receptor‐associated protein (GABARAP), GABA type A receptor‐associated protein‐like 1 (GABARAPL1), and GABA type A receptor‐associated protein‐like 2 (GABARAPL2; best known as GATE‐16) (Shpilka et al, 2011) (Table 1).
Throughout the past decade, the terms “canonical” and “non‐canonical” have been extensively employed to (i) refer to non‐degradative functions of macroautophagy (e.g., unconventional secretion) (Ponpuak et al, 2015), or (ii) discriminate between those macroautophagic responses that critically rely on ATG3‐, ATG5‐, ATG7‐, ULK1‐, BECN1‐, and VPS34‐mediated PI3P production and those that do not (Codogno et al, 2012; Ktistakis & Tooze, 2016). Although this latter use of the adjectives “canonical” and “non‐canonical” may be advantageous as it refers to molecular signatures that are shared by various instances of macroautophagy, we fear that it might be rather misleading, for at least two reasons. First, they implicitly convey the notion that some macroautophagic responses are frequent and observable in many distinct experimental settings, while others are relatively exceptional. The literature describes hundreds of scenarios in which macroautophagy can be slowed down by the inhibition of ATG3‐, ATG5‐, ATG7‐, ULK1‐, BECN1‐, and VPS34‐dependent PI3P production, but only a few instances of ATG3‐, ATG5‐, ATG7‐, ULK1‐, BECN1‐, VPS34,‐ and PI3P‐independent macroautophagic responses (Nishida et al, 2009; Niso‐Santano et al, 2015; Vicinanza et al, 2015). However, this imbalance might stem from an observational bias linked to the stimuli used to elicit autophagy (starvation, rapamycin or targeted cellular damage) and/or to the biomarkers used so far to monitor macroautophagic responses (such as LC3 lipidation) (Klionsky et al, 2016). Second, and perhaps most important, a real consensus on the set of features that would characterize “canonical” versus “non‐canonical” macroautophagy has never been reached. Thus, while some authors have used the term “non‐canonical” for ATG5‐dependent, BECN1‐independent cases of macroautophagy (Niso‐Santano et al, 2015; Huang & Liu, 2016), others have employed the same expression for ULK1‐independent, ATG5‐ and BECN1‐dependent macroautophagic responses (Martinez et al, 2016). To avoid confusion, we propose to avoid terms such as “canonical” and “non‐canonical”. Rather, we encourage the use of explicit expressions such as “ATG5‐dependent”, “BECN1‐independent” and alike, provided that such a dependence/independence has been experimentally verified. Of note, this recommendation does not intend to imply the existence of distinct pathways that fully depend or not on specific components of the macroautophagy apparatus, but to support the description of a specific instance of macroautophagy based on experimental validation.
As for the definition of macroautophagic responses, relying upon specific components of the underlying molecular apparatus may also be relatively misleading. We propose therefore a functional definition of macroautophagy as a type of autophagic response (i.e., a response that involves the lysosomal degradation of a cytosolic entity, see above) that relies on autophagosomes, which can be subtyped based upon dependence on specific proteins. Comprehensive guidelines provide robust methods to monitor the formation of functional autophagosomes and autophagic flux (Klionsky et al, 2016). We surmise that a common molecular signature of macroautophagic responses may be difficult to identify, at least in part owing to the high degree of redundancy and interconnectivity of the process (at least in mammalian cells).
Non‐selective and selective types of autophagy
Micro‐ and macroautophagic responses can involve disposable cytoplasmic components in a relatively non‐selective manner. Upon lysosomal degradation, these autophagy substrates fuel bioenergetic metabolism or repair processes (Liu et al, 2015a; Sica et al, 2015). In addition, microautophagy, macroautophagy, and CMA can operate in a specific manner, through a mechanism that involves the recognition of autophagy substrates by dedicated receptors (Farre & Subramani, 2016). In this setting, it is useful to remember that the specificity of autophagic responses is highly affected by the mechanisms of substrate delivery to lysosomes. Thus, whereas CMA appears as a highly selective type of autophagy (as it virtually operates only on cytosolic proteins containing KFERQ‐like motives bound to HSPA8 and compatible with LAMP2A‐mediated translocation), both microautophagy and macroautophagy can exhibit incomplete specificity under specific conditions (reflecting the relatively “leaky” processes of lysosomal invagination and autophagosome formation, respectively) (Sica et al, 2015; Zaffagnini & Martens, 2016). This notion should be kept under attentive consideration when specific instances of autophagy (see below) are measured. The literature offers a collection of articles in which specificity was not addressed, as investigators focused on the degradation of a single substrate (e.g., damaged mitochondria) but did not monitor to which extent other cytoplasmic entities were also degraded. Thus, it may be difficult to differentiate between non‐selective micro‐ or macroautophagic responses and their specific counterparts, especially for some substrates like mitochondria. Indeed, mitophagy (see below for a definition) is arguably the best‐characterized form of selective macroautophagy (at least in mammalian cells), but parts of the mitochondrial network are also degraded in the course of macroautophagic responses driven by bioenergetic needs (Gomes et al, 2011a,b). We propose to define specific instances of micro‐ and macroautophagy based on the enrichment of a precise autophagy substrate, coupled to requirement of specific molecular factors (such as autophagy receptors), which may be used to selectively monitor or experimentally manipulate the process (Table 1).
Mitophagy
Mitophagy can be defined as the specific removal of damaged or excess mitochondria by micro‐ or macroautophagy. Microautophagic responses preferentially targeting mitochondria have been observed in yeast cells submitted to nitrogen starvation (Kissova et al, 2007). In this system, the microautophagic response depends on SUN family protein UTH1 (Uth1), an integral factor of the inner mitochondrial membrane (Kissova et al, 2007). Whether Uth1 is the actual receptor for mitochondrial microautophagy, however, remains to be determined. Conversely, macroautophagic responses specific for mitochondria have been described in a wide panel of model organisms, including yeast, nematodes, flies, and mammals. This process contributes to the removal of superfluous mitochondria that have no functional defects a priori, as well as to the degradation of mitochondria that are damaged beyond repair, hence dysfunctional and potentially cytotoxic (which is critical for the maintenance of cellular homeostasis, especially in highly metabolic tissues such as the brain) (Palikaras & Tavernarakis, 2014). Two physiological settings exemplify the macroautophagic removal of functional mitochondria: (i) the maturation of reticulocytes and consequent formation of mature erythrocytes, a setting in which mitophagy critically relies on BCL2 interacting protein 3‐like (BNIP3L; best known as NIX) and the complete removal of mitochondria may also depend on unconventional secretion (Sandoval et al, 2008; Mortensen et al, 2010; Novak et al, 2010; Griffiths et al, 2012; Fader et al, 2016); (ii) the first steps of embryonic development (Al Rawi et al, 2011; Sato & Sato, 2011), in which paternal mitochondria undergo fission, mitochondrial 1 (FIS1)‐dependent fragmentation (Rojansky et al, 2016; Wang et al, 2016), lose transmembrane potential (Rojansky et al, 2016; Wang et al, 2016) and are removed by a mitophagic response depending on endonuclease G (ENDOG; at least in Caenorhabditis elegans) (Zhou et al, 2016), prohibitin 2 (PHB2) (Wei et al, 2017), PTEN‐induced putative kinase 1 (PINK1), and Parkinson disease (autosomal recessive, juvenile) 2, parkin (PARK2) (in mammals, but not in D. melanogaster) (Politi et al, 2014; Rojansky et al, 2016). In this scenario, CPS‐6 (the worm orthologue of ENDOG) promotes mitophagy via a poorly characterized mechanism that involves the degradation of the mitochondrial genome (Zhou et al, 2016), whereas PHB2 and the PINK1‐PARK2 system contribute to the generation of tags recognizable by LC3 or autophagy receptors, respectively (Geisler et al, 2010; Narendra et al, 2010; Wei et al, 2017).
The selective removal of depolarized mitochondria also involves the PINK1‐PARK2 system and PHB2 (Clark et al, 2006; Park et al, 2006), which generate ubiquitin and non‐ubiquitin tags at damaged mitochondrial membranes to allow recognition by sequestosome 1 (SQSTM1, best known as p62) (to a limited extent), optineurin (OPTN), calcium binding and coiled‐coil domain 2 (CALCOCO2; best known as NDP52), and LC3 (Wong & Holzbaur, 2014; Heo et al, 2015; Lazarou et al, 2015; Moore & Holzbaur, 2016; Wei et al, 2017). Cardiolipin, a mitochondrial lipid, has also been proposed to directly interact with LC3 upon mitochondrial damage caused by a variety of stimuli (Chu et al, 2013; Kagan et al, 2016). FUN14 domain containing 1 (FUNDC1), a protein of the outer mitochondrial membrane, operates as autophagy receptor in response to hypoxia (Liu et al, 2012). Finally, SMAD‐specific E3 ubiquitin‐protein ligase 1 (SMURF1), peroxisomal biogenesis factor 3 (PEX3), PEX13, various members of the Fanconi anemia (FA) protein family and transglutaminase 2 (TGM2) have also been involved in the regulation or execution of mitophagy, although their exact role remains to be elucidated (Orvedahl et al, 2011; Rossin et al, 2015; Lee et al, 2017; Sumpter et al, 2016). Atg32 is the main receptor for macroautophagic responses targeting dispensable mitochondria in yeast (Kanki et al, 2009; Okamoto et al, 2009), and BCL2‐like 13 (BCL2L13) has been suggested to play analogous functions in mitophagy in mouse and human cells (Murakawa et al, 2015). In C. elegans, macroautophagic responses specific for mitochondria are coordinated with mitochondrial biogenesis owing to the coordinated activity of the BNIP3 homologue DCT‐1 and the transcription factor SNK‐1 (Palikaras et al, 2015).
Pexophagy
Pexophagy is a macroautophagic response preferentially targeting peroxisomes. In yeast, a large supramolecular complex is responsible for the selective recognition of peroxisomes by the molecular machinery for macroautophagy and their actin‐dependent transport to the vacuole (Reggiori et al, 2005). This complex includes the peroxisomal proteins Pex3 (Burnett et al, 2015), Pex14 (Zutphen et al, 2008) as well as Atg37 (Nazarko et al, 2014), which are bound by Atg30 (Burnett et al, 2015), Atg11 (Burnett et al, 2015; Torggler et al, 2016), and Atg36 (Motley et al, 2012; Tanaka et al, 2014). In mammalian cells, pexophagy proceeds upon the PEX2‐ and PEX3‐dependent ubiquitination of multiple peroxisomal proteins including PEX5 and ATP‐binding cassette subfamily D member 3 (ABCD3; best known as PMP70), which are recognized by the autophagy receptors p62 and NBR1 (Deosaran et al, 2013; Yamashita et al, 2014; Sargent et al, 2016). Mammalian pexophagy is highly responsive to oxidative stress, possibly as a consequence of cytoplasmic ATM activation or endothelial PAS domain protein 1 (EPAS1; best known as HIF‐2α) signaling (Walter et al, 2014; Zhang et al, 2015). Of note, the selective degradation of peroxisomes in yeast has also been shown to occur through a selective form of microautophagy termed micropexophagy (Farre & Subramani, 2004).
Nucleophagy
Nucleophagy can be defined as an autophagic response selectively targeting portions of the nucleus. In yeast, two distinct forms of nucleophagy have been described: (i) a microautophagic form that relies on the autophagy receptor Nvj1, the vacuolar protein Vac8 and members of the oxysterol‐binding protein (OSBP) family (Roberts et al, 2003; Kvam & Goldfarb, 2004), which has been dubbed “piecemeal microautophagy of the nucleus”; and (ii) a variant that does not require Nvj1, Vac8 but does involve components of the macroautophagy machinery, such as Atg3 and Atg4 (but not Atg6, the yeast orthologue of BECN1) (Krick et al, 2008; Mijaljica et al, 2012), and the autophagy receptor Atg39 (Mochida et al, 2015). Nucleophagy also occurs in mammalian cells (Park et al, 2009), in which it contributes to the maintenance of genomic integrity (Rello‐Varona et al, 2012; Dou et al, 2015). Lamin B1 (LMNB1) has been identified as the nuclear protein responsible for a variant of nucleophagy in mammalian cells (Dou et al, 2015).
Reticulophagy
Reticulophagy is the preferential autophagic degradation of portions of the ER. According to some authors, reticulophagy (also called ER‐phagy) occurs independently of both the micro‐ and macroautophagy machinery, at least in yeast (Schuck et al, 2014), but is regulated by the Rab family GTPase Ypt1 (Lipatova et al, 2013). Other authors, however, provided evidence suggesting that reticulophagy constitutes a specific form of macroautophagy that relies on the autophagy receptors Atg39 and Atg40 (in yeast), or their mammalian orthologue family with sequence similarity 134 member B (FAM134B) (in human and mouse cells) (Khaminets et al, 2015; Mochida et al, 2015). In S. cerevisiae, reticulophagy also involves Atg11 (Mochida et al, 2015) and Sec63 complex subunit SEC62 (Sec62) (Fumagalli et al, 2016).
Ribophagy
Ribophagy is a specific autophagic response targeting ribosomes. In yeast, ribophagy involves ribosomal de‐ubiquitination by the mRNA‐binding ubiquitin‐specific protease Ubp3 and its cofactors Bre5, Doa1 (also known as Ufd3), and Cdc48 (Kraft et al, 2008; Ossareh‐Nazari et al, 2010) and requires Atg11 (Waliullah et al, 2017). Conversely, the autophagic removal of dispensable ribosomes is negatively regulated by listerin E3 ubiquitin‐protein ligase 1 (Ltn1)‐dependent ubiquitination (Ossareh‐Nazari et al, 2014), and possibly by NEDD4 family E3 ubiquitin‐protein ligase Rsp5 (Shcherbik & Pestov, 2011). Ubp3 has also been involved in the autophagic and proteasomal removal of translation and RNA turnover factors during nitrogen starvation (Kelly & Bedwell, 2015). Ribophagy driven by nutrient starvation in yeast is accompanied by bulk RNA degradation within the vacuole (Huang et al, 2015). Interestingly, some plants exhibit a microautophagic variant of ribophagy (Niki et al, 2014). To the best of our knowledge, ribophagic responses in mammalian cells have not yet been described.
Aggrephagy
Aggrephagy can be defined as an autophagic response specific for protein aggregates. Aggrephagy has been described in a variety of model organisms, including yeast (Lu et al, 2014b), worms (Jia et al, 2007; Lu et al, 2013), flies (Simonsen et al, 2008), plants (Toyooka et al, 2006), and mammals (Bjorkoy et al, 2005; Hara et al, 2006; Komatsu et al, 2006). The macroautophagic disposal of protein aggregates is particularly relevant for the preservation of cellular homeostasis, especially in the context of neurodegenerative disorders (Menzies et al, 2015). Besides relying on the macroautophagy machinery and often on substrate ubiquitination, mammalian aggrephagy involves the autophagy receptors p62 (which can form insoluble aggregates itself) (Bjorkoy et al, 2005; Komatsu et al, 2007; Pankiv et al, 2007; Kirkin et al, 2009b), NBR1 (an orthologue of which participates in plant aggrephagy) (Kirkin et al, 2009a,b), OPTN (Korac et al, 2013), and toll‐interacting protein (TOLLIP) (Lu et al, 2014b), as well as the p62‐binding proteins WD repeat and FYVE domain containing 3 (WDFY3; best known as ALFY) (Simonsen et al, 2004; Filimonenko et al, 2010) and TGM2 (D'Eletto et al, 2012). However, it is worth noting that the redundancy between these factors and their specific roles in the degradation of different substrates has not been extensively explored. In yeast, the ubiquitin‐binding protein Cue5 (the orthologue of mammalian TOLLIP) operates as autophagy receptor for aggrephagic responses (Lu et al, 2014b). In D. melanogaster the control of proteostasis by aggrephagy impinges on forkhead box, subgroup O (FOXO)‐dependent transcription (Demontis & Perrimon, 2010). Importantly, LC3 can accumulate at protein aggregates in a p62‐dependent but autophagosome‐independent manner (Kuma et al, 2007; Shvets & Elazar, 2008). This adds to the potential sources of bias deriving from the use of GFP‐LC3 aggregation as a standalone biomarker for macroautophagy (see above). HSPA8 as well as other chaperones and co‐chaperones have been involved in a specific form of aggrephagy commonly known as “chaperone‐assisted selective autophagy” (CASA) (Arndt et al, 2010). CASA differs from endosomal microautophagy and CMA in its dependence on multiple components of the macroautophagy apparatus, de facto constituting a selective form of macroautophagy (Arndt et al, 2010).
Lipophagy
Lipophagy is the selective autophagic degradation of neutral lipid droplets. Originally discovered in the mammalian system, where it involves the molecular machinery for macroautophagy (Singh et al, 2009), lipophagy also occurs in worms and in yeast. In C. elegans, lipophagy involves lysosomal lipases such as LIPL‐4, which play key signaling roles in longevity (Lapierre et al, 2011; O'Rourke & Ruvkun, 2013; Folick et al, 2015). In yeast, it involves a microautophagic process (Wang et al, 2014; Vevea et al, 2015). However, there are contradicting reports on the molecular requirements for S. cerevisiae lipophagic responses to intracellular lipid accumulation (Wang et al, 2014; Vevea et al, 2015). Thus, while some authors propose that lipophagy in yeast does not involve Atg7 but requires ESCRT components (Vevea et al, 2015), other authors favor the interpretation that lipophagic responses in yeast depend on Atg7 and several other components of the macroautophagy machinery (even though they manifest with a microautophagic appearance and proceed in the absence of autophagosomes) (Wang et al, 2014). In mammalian cells, lipophagy is coordinated by transcriptional programs depending on nuclear receptor subfamily 1 group H member 4 (NR1H4; best known as FXR), cAMP responsive element binding protein 1 (CREB), and peroxisome proliferator‐activated receptor alpha (PPARA) (Lee et al, 2014; Seok et al, 2014). Interestingly, the CMA‐dependent degradation of lipid droplet‐associated proteins such as perilipin 2 (PLIN2) and PLIN3 precedes and facilitates lipolysis (Kaushik & Cuervo, 2015, 2016), demonstrating the existence of intimate cross talk between different forms of autophagy in the control of intracellular homeostasis. Moreover, several autophagy genes including bec‐1 (the worm orthologue of BECN1) are required for the accumulation of neutral lipids in the intestine of developing C. elegans (Lapierre et al, 2013), pointing to a broader implication of autophagy in systemic lipid homeostasis.
Bacterial xenophagy
Bacterial xenophagy is the macroautophagic removal of cytoplasmic bacteria, that is, bacteria that escape the phagosomal compartment upon phagocytosis, and damaged bacteria‐containing phagosomes. As mentioned above, bacterial xenophagy must be conceptually discriminated from efficient phagocytosis, a setting in which bacteria never gain direct access to the cytosolic milieu (Huang & Brumell, 2014). Xenophagic responses targeting bacteria constitute a first, cell‐autonomous line of innate defense against prokaryotic infections (Deretic et al, 2013). Accordingly, multiple bacteria have evolved strategies to actively inhibit autophagic responses in the host (Galluzzi et al, 2017b). In mammalian cells, cytoplasmic bacteria are rapidly recognized by various autophagy receptors including p62, OPTN, NDP52, and Tax1‐binding protein 1 (TAX1BP1), via a mechanism that relies on receptor phosphorylation by TANK1‐binding kinase 1 (TBK1) (Thurston et al, 2009; Wild et al, 2011; Tumbarello et al, 2015) and ubiquitination by ring finger protein 166 (RNF166) (Heath et al, 2016). Additional proteins that direct the formation and expansion of autophagosomes to sites of bacterial invasions include (but may not be limited to) WD repeat domain, phosphoinositide interacting 2 (WIPI2), and its interactor TECPR1, which are recruited in a TBK1‐dependent manner (Ogawa et al, 2011; Thurston et al, 2016), as well as the pattern recognition receptors nucleotide‐binding oligomerization domain containing 1 (NOD1) and NOD2, which physically interact with ATG16L1 and immunity‐related GTPase M (IRGM) upon recognition of bacterial muramyl dipeptide (Cooney et al, 2010; Travassos et al, 2010; Chauhan et al, 2015). Besides operating as a receptor for the recruitment of forming autophagosomes to invading bacteria, NDP52 supports autophagosome maturation upon interaction with LC3A, LC3B, LC3C, GABARAPL2, and myosin VI (MYO6) (von Muhlinen et al, 2012; Verlhac et al, 2015). Ubiquitin D (UBD; best known as FAT10) has also been involved in the rapid and transient recognition of phagosome‐escaping bacteria, and FAT10 deficiency has been associated with increased susceptibility to Salmonella typhimurium infection in mice (Spinnenhirn et al, 2014). The molecular mechanisms through which FAT10 supports xenophagy, however, remain to be clarified. Interestingly, xenophagic responses targeting damaged phagosomes and their bacterial cargo have been described. This particular variant of xenophagy relies on galectin 8 (LGALS8) or galectin 3 (LGALS3), both of which tag damaged endosomes (Chauhan et al, 2016), as well as on NDP52 (Thurston et al, 2012; Kim et al, 2013a; Li et al, 2013) and/or various members of the TRIM protein family as receptors or receptor regulators (see below for a definition) (Kimura et al, 2015, 2016). Although xenophagic responses have mainly been studied in the mammalian system, there are bona fide instances of xenophagy in D. melanogaster, in which it also operates at the boundary of innate pattern recognition (Wu et al, 2007; Yano et al, 2008; Kim et al, 2012), C. elegans (Jia et al, 2009; Zou et al, 2014) and Dictyostelium discoideum (Jia et al, 2009).
Viral xenophagy
Viral xenophagy (virophagy) is a macroautophagic response targeting fully formed cytoplasmic virions or components thereof. The first description of endogenous membranes engulfing cytoplasmic viruses dates back to the late 1990s (Schlegel et al, 1996), and it is now clear that virophagy occupies a position similar to that of bacterial xenophagy in the first line of defense against pathogens (Paul & Munz, 2016). In line with this notion, several defects in the molecular machinery for macroautophagy—such as the genetic inhibition of Atg5 in mice—render animals more susceptible to succumb to infection (Orvedahl et al, 2010). This holds true not only in mammalian systems, but also in plants (Liu et al, 2005), flies (Nakamoto et al, 2012; Moy et al, 2014) and perhaps nematodes (Bakowski et al, 2014). Moreover, HIV‐1+ patients who remain clinically stable for years in the absence of therapy (so‐called long‐term non‐progressors) display high baseline levels of autophagy in peripheral blood mononuclear cells (Nardacci et al, 2014). Accordingly, multiple viruses have evolved strategies to avoid host virophagic responses, including the expression of BECN1 inhibitors (Orvedahl et al, 2007; Levine et al, 2011) or proteins that inhibit the autophagosomal‐lysosomal fusion (Gannage et al, 2009). Besides relying on the core macroautophagy machinery, efficient virophagic responses involve p62 and tripartite motif containing 5 (TRIM5) as receptors (Orvedahl et al, 2010; Mandell et al, 2014), proteins that participate in mitophagy, such as SMURF1 (Orvedahl et al, 2011), Fanconi anemia complementation group C (FANCC) (Sumpter et al, 2016), and PEX13 (Lee et al, 2017), as well as the phosphorylation of eukaryotic translation initiation factor 2A (EIF2A) (Talloczy et al, 2002).
Proteaphagy
Proteaphagy is a term coined to indicate macroautophagic responses specific for inactive proteasomes. In Arabidopsis thaliana, proteaphagy relies on the proteasomal component regulatory particle non‐ATPase 10 (RPN10), which operates as a bona fide autophagy receptor to bridge ubiquitinated proteasome subunits to ATG8 (Marshall et al, 2015). In yeast, Rpn10 is dispensable for proteaphagy (Waite et al, 2016) but a similar function is mediated by Cue5 (Marshall et al, 2016), drawing an interesting parallelism with aggrephagy (see above). Besides involving Atg7, optimal proteaphagic responses in S. cerevisiae rely on the co‐chaperone Hsp42 (Marshall et al, 2016). Thus, it is tempting to speculate that the macroautophagic disposal of inactive proteasomes may proceed upon their accumulation in aggregates, at least in yeast. Mammalian cells subjected to starvation and other stressful conditions mount proteaphagic responses that mainly on p62 as a receptor (Cuervo et al, 1995; Cohen‐Kaplan et al, 2016).
Lysophagy
Lysophagy is the specific macroautophagic disposal of damaged lysosomes in mammalian cells. Several lysosomotropic agents as well as monosodium urate (MSU) and silica have been shown to promote lysosomal damage followed by ubiquitination and recruitment of the macroautophagy machinery (Hung et al, 2013; Maejima et al, 2013), a process that may be directed by the common marker of endovesicular damage LGALS3 (Kawabata & Yoshimori, 2016). Most of the molecular details underlying lysophagy, however, remain to be determined. Similarly, if and how a lysophagy‐like mechanism contributes to the preservation of vacuolar homeostasis in yeast and plants remains obscure.
Other specific forms of autophagy
Additional instances of selective macroautophagy have been described, mostly based on cargo selectivity. These include (but are likely not limited to): myelinophagy (targeting myelin in Schwann cells) (Gomez‐Sanchez et al, 2015), zymophagy (targeting zymogen granules in pancreatic acinar cells) (Grasso et al, 2011), granulophagy (targeting stress granules) (Buchan et al, 2013), and ferritinophagy (targeting ferritin via the receptor nuclear receptor coactivator 4, NCOA4) (Dowdle et al, 2014; Mancias et al, 2014). Finally, macroautophagy has been involved in the degradation of specific proteins owing to their ability to physically interact with members of the Atg8 protein family. This applies, for instance, to the centriole and centriolar satellite protein OFD1, whose degradation by macroautophagy has a major impact on the regulation of ciliogenesis (Tang et al, 2013). A term to indicate such a protein‐specific variant of macroautophagy has yet to be proposed.
Autophagic flux
All forms of autophagy are multistep processes during which autophagy substrates are recognized, isolated (biochemically and/or physically) from the cytoplasmic milieu, and delivered to lysosomes for degradation. In physiological conditions, microautophagy, CMA, and macroautophagy proceed at baseline levels, hence contributing to the preservation of cellular homeostasis as they avoid the accumulation of potentially cytotoxic entities that may accumulate as a result of normal cellular functions (e.g., damaged mitochondria) (Li et al, 2012; Cuervo & Wong, 2014; Sica et al, 2015). In addition, all autophagic pathways described so far are sensitive to perturbations of intracellular or extracellular homeostasis. Thus, stimuli as different as nutritional, metabolic, chemical, physical, and hormonal cues can alter (increase or decrease) the ability of microautophagy, CMA, and macroautophagy to degrade autophagy substrates (Galluzzi et al, 2014; Green & Levine, 2014; Kaur & Debnath, 2015; Mukherjee et al, 2016; Tasset & Cuervo, 2016). The rate at which lysosomes degrade autophagy substrates is a good indicator of such a global efficiency in autophagic responses, which is commonly known as “autophagic flux” (Loos et al, 2014). The importance of this concept leaps to the eye upon considering macroautophagic responses and some of the biomarkers that have been employed so far to measure them, such as LC3 lipidation (as monitored by immunoblotting) and the formation of GFP‐LC3+ cytoplasmic dots (as monitored by immunofluorescence microscopy) (Klionsky et al, 2016). Both LC3 lipidation and GFP‐LC3+ cytoplasmic dots, indeed, are relatively reliable indicators of the pool size of the autophagosomal compartment, which is known to expand in the course of productive macroautophagic responses (increased on‐rate) (Klionsky et al, 2016). However, autophagosomes also accumulate when the formation of autolysosomes or lysosomal degradation is blocked (decreased off‐rate), a situation in which autophagy substrates are not disposed of (Boya et al, 2005; Gonzalez‐Polo et al, 2005). Moreover, it cannot be excluded that the autophagosomal compartment also mediates autophagy‐independent functions. Although several techniques are currently available to monitor autophagic flux in real time (Katayama et al, 2011; Kaizuka et al, 2016) and to discriminate between situations of increased on‐rate and situations of decreased off‐rate (Klionsky et al, 2016), this profound difference should be kept under critical consideration. In summary, the term autophagic flux refers to the rate at which the molecular machinery for autophagy identifies, segregates, and disposes of its substrates (through lysosomal degradation).
Autophagy‐dependent cell death
Since the very beginning of the field, when microscopy was the main (if not the sole) experimental approach for the study of cell biology, scientists have been observing cells that die as they accumulate autophagosomes and autolysosomes in the cytoplasm (Schweichel & Merker, 1973; Eskelinen et al, 2011). Morphologically, these cells differ considerably from cells undergoing apoptosis or necrosis (be it regulated or accidental), which led investigators to adopt the term “autophagic cell death” or “type II cell death” based on observational/correlational (rather than interventional/causal) grounds (Schweichel & Merker, 1973; Kroemer et al, 2009). With the advent of modern molecular biology, it has become clear that macroautophagy has robust cytoprotective functions in the majority of pathophysiological and experimental settings (Menzies et al, 2015; Galluzzi et al, 2016). Indeed, pharmacological inhibitors of macroautophagy as well as genetic interventions targeting various components of the macroautophagy machinery generally accelerate (rather than retard) the demise of cells experiencing perturbations of homeostasis (Boya et al, 2005; Yousefi et al, 2006; Mrschtik et al, 2015). Thus, RCD often occurs in the context of failing macroautophagic responses that are activated as an ultimate attempt of the cell to preserve homeostasis (Galluzzi et al, 2015a).
Importantly, there are numerous exceptions to this tendency, suggesting that functional macroautophagic responses or components of the machinery for macroautophagy can also: (i) have little, if any, impact on RCD (so‐called non‐protective autophagy) (Saleh et al, 2016); or (ii) etiologically contribute to RCD (at least in specific developmental or pathophysiological scenarios) (Seay & Dinesh‐Kumar, 2005; Masini et al, 2009; Sharma et al, 2014; Denton et al, 2015). For instance, disrupting any of several Atg genes in D. melanogaster, as well as blocking autophagy initiation by modulating growth signaling, results in a failure to remove larval salivary gland and midgut tissue during metamorphosis (Berry & Baehrecke, 2007; Denton et al, 2009, 2013; Xu et al, 2015). Interestingly, larval midgut degradation, which occurs independent of caspase‐dependent apoptosis, does not require all components of the macroautophagy apparatus involved in starvation‐induced autophagy in the Drosophila fat body (Xu et al, 2015).
Moreover, pharmacological and genetic data indicate that a specific form of autophagy‐dependent cell death involving the plasma membrane Na+/K+‐ATPase (called “autosis”) occurs in cells exposed to nutrient deprivation or a BECN1‐derived peptide, as well as in the brain of newborn rodents experiencing ischemia/hypoxia (Liu et al, 2013; Xie et al, 2016). In summary, autophagy‐dependent cell death can be defined as a form of RCD that can be retarded by pharmacological or genetic inhibition of macroautophagy. In this context, it is important to note that (i) specificity issues affect most, if not all, pharmacological agents employed so far for suppressing macroautophagic responses (Maycotte et al, 2012; Maes et al, 2014; Eng et al, 2016; Galluzzi et al, 2017b); and (ii) multiple components of the macroautophagy machinery have autophagy‐independent functions (Hwang et al, 2012; Maskey et al, 2013). Thus, we recommend to favor genetic approaches and to test the involvement of at least two different proteins of the macroautophagy apparatus in a specific instance of RCD before etiologically attributing it to macroautophagy. Expressions such as “ATG5‐dependent cell death” or “BECN1‐dependent cell death” may be even more appropriate when the involvement of one or more specific components of the macroautophagy apparatus has been experimentally validated in the absence of links to increased autophagic flux. Autosis can be functionally defined as a Na+/K+‐ATPase‐mediated form of autophagy‐dependent cell death.
Cytoplasm‐to‐vacuole targeting pathway
The cytoplasm‐to‐vacuole targeting (Cvt) pathway delivers hydrolases including aminopeptidase 1 (Ape1), Ape4, and alpha‐mannosidase (Ams1) to the yeast vacuole (Umekawa & Klionsky, 2012). The molecular machineries for the Cvt pathway and macroautophagy share a large number of components, including several Atg proteins (Scott et al, 1996, 2000, 2001). Moreover, Ape1, Ape4, and Ams1 are imported into the vacuole as large oligomers, being reminiscent of the substrates of aggrephagy (Bertipaglia et al, 2016). The Cvt pathway, however, contributes to the preservation of normal enzymatic activity within the vacuole, especially in vegetative conditions, de facto mediating biosynthetic, rather than catabolic, functions (Umekawa & Klionsky, 2012). Thus, the Cvt pathway does not represent an instance of autophagy strictly speaking.
LC3‐associated phagocytosis
LC3‐associated phagocytosis (LAP) describes the recruitment of some (but not all) components of the macroautophagy apparatus (notably, LC3) to single‐membraned phagosomes that contain extracellular pathogens or dead cell corpses destined to lysosomal degradation (Sanjuan et al, 2007; Martinez et al, 2015, 2016). Multiple molecular determinants of LAP are also required for macroautophagic responses. This applies to ATG3, ATG5, ATG7, ATG12, ATG16L1, BECN1, VPS34, and UVRAG (Martinez et al, 2015, 2016). However, in the mammalian systems investigated thus far, LAP does not involve ULK1 signaling, AMBRA1 and ATG14 (which are also involved in macroautophagy), but critically depends on RUBICON and NAPDH oxidase 2 (which are dispensable for macroautophagy). LAP has been involved in the control of bacterial and fungal pathogens (Sanjuan et al, 2007; Zhao et al, 2008; Gong et al, 2011; Lam et al, 2013; Choi et al, 2014; Martinez et al, 2015; Selleck et al, 2015), in entosis (a variant of RCD that ensues engulfment by non‐phagocytic cells) (Florey et al, 2011), as well as in the optimal disposal of dead cells (Martinez et al, 2016). However, since the substrates of LAP are extracellular entities that never enter the cytoplasm, LAP cannot be considered as a bona fide autophagic response.
Secretory autophagy
Multiple components of the molecular apparatus for macroautophagy including (but presumably not limited to) ATG4B, ATG5, ATG7, ATG16L1, BECN1, ULK1, LC3, p62, some SNAREs and specific members of the TRIM protein family also participate in the conventional or unconventional secretion of cytoplasmic entities (including soluble proteins with extracellular functions, potentially cytotoxic protein aggregates, secretory granules, and invading pathogens) (Manjithaya et al, 2010; Dupont et al, 2011; Shravage et al, 2013; Lock et al, 2014; Gerstenmaier et al, 2015; Kimura et al, 2017), which led to the introduction of the term “secretory autophagy” (Ponpuak et al, 2015). Although these non‐degradative functions of the macroautophagy machinery are essential for multiple intracellular and organismal processes, including viral clearance, inflammation, and hematopoiesis, they should not be considered as bona fide autophagic responses. Along these lines, we encourage the use of molecularly oriented expressions such as “ATG5‐dependent secretion” over potentially misleading terms including “secretory autophagy”.
Crinophagy
The term crinophagy refers to the degradation of secretory material following the fusion of secretory granules with lysosomes (Marzella et al, 1981). This process, which has been observed in secretory cells and is distinct from zymophagy, ensures the degradation and recycling of excess/obsolete secretory granules, for instance, those that persist after a hormone‐induced wave of secretion is over (Weckman et al, 2014). Strictly speaking, crinophagy should not be considered as a form of autophagy as the content of secretory granules is not accessible from the cytoplasm (it is contained in secretory granules, similar to endosomal or phagosomal cargoes).
Components of the autophagy machinery
Autophagy substrates (autophagic cargo)
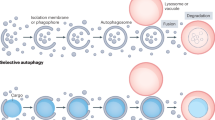
The terms autophagy substrates and autophagic cargo can be interchangeably used to describe a large and heterogeneous set of cytoplasmic entities (of endogenous or exogenous origin) that are targeted to lysosomal degradation by autophagy (Fig 1). From a conceptual standpoint, autophagy substrates should be differentiated from autophagy receptors (see below). Indeed, both autophagy substrates and receptors are subjected to lysosomal degradation, but only the latter function as part of the autophagy apparatus (Boya et al, 2013; Noda & Inagaki, 2015; Zaffagnini & Martens, 2016). Of note, neither hydrolytic enzymes delivered to the vacuole via Cvt (which contribute to the preservation of enzymatic homeostasis) nor extracellular entities reaching lysosomes via the endocytic pathway (which never enter the cytoplasm) can be considered as bona fide autophagy substrates.

Autophagy substrates
A wide and heterogeneous set of cytoplasmic entities—be they of endogenous/intracellular or exogenous/extracellular origin—can be targeted to lysosomal degradation by non‐selective or selective forms of autophagy. ER, endoplasmic reticulum.
Autophagy receptors and adaptors
An autophagy receptor is any of the proteins that bind autophagy substrates, allow for their recognition by the autophagy machinery, and become degraded within lysosomes in the course of functional autophagic responses (Stolz et al, 2014). Based on this definition, HSPA8 is the main receptor for endosomal microautophagy but not for CMA (during CMA, the cytoplasmic pool of HSPA8 is not degraded) (Uytterhoeven et al, 2015; Morozova et al, 2016). In addition, dozens of proteins have been involved in the recognition of macroautophagy substrates (see above) (Rogov et al, 2014; Farre & Subramani, 2016). Most receptors for macroautophagy share an evolutionary conserved LC3‐interacting region (LIR), which allows them to bring macroautophagy substrates in the proximity of LC3+ forming autophagosomes. This applies to p62, NBR1, OPTN, NDP52, BNIP3, BNIP3L, ATG34, FUNDC1, PHB2, TRIM5, TAX1BP1, Atg19, and Atg32 (Birgisdottir et al, 2013; Chourasia et al, 2015; Wei et al, 2017). Many macroautophagy receptors also contain ubiquitin‐binding domains, allowing them to recruit ubiquitinated substrates to forming autophagosomes (Khaminets et al, 2016). Moreover, some receptors including yeast Atg19 and Atg34 as well as human p62, OPTN, and NDP52 have been shown to bind to the Atg12‐Atg5:Atg16 (ATG12‐ATG5:ATG16L1) complex to stimulate conjugation of Atg8 family members at the autophagic cargo (Fracchiolla et al, 2016). Along similar lines, multiple members of the TRIM protein family not only target autophagy substrates to forming autophagosomes upon LC3 binding, but also physically and functionally interact with upstream components of the autophagy apparatus, including the ULK1 and VPS34 complexes (Kimura et al, 2015, 2016). These proteins have been dubbed “receptor regulators”. It cannot be excluded that other autophagy receptors might have regulatory functions besides cargo recognition.
Although the term autophagy adaptor has also been used as a synonym of autophagy receptor, we recommend to employ this expression to indicate any of the proteins that interact with Atg8 family members but are not involved in cargo recognition (and hence not degraded during macroautophagic responses) (Stolz et al, 2014). Two examples of autophagy adaptors outside of the ATG protein family (many members de facto behave as adaptors) are FYVE and coiled‐coil domain containing 1 (FYCO1), which is involved in the interaction of autophagosomes with the cytoskeleton and their fusion with lysosomes, and sorting nexin 18 (SNX18), which participates in autophagosome formation (Knaevelsrud et al, 2013; Olsvik et al, 2015).
Phagophores (isolation membranes)
Phagophores (also called isolation membranes) are the precursors of autophagosomes. Mammalian phagophores generally form near ER‐mitochondria contact sites in the context of unique structures staining positively for zinc finger FYVE‐type containing 1 (ZFYVE1; best known as DFCP1) known as omegasomes (Axe et al, 2008). In mammals, phagophore biogenesis has been suggested to involve ATG9‐containing vesicles that derive from the Golgi apparatus, late endosomes or the plasma membrane (Ravikumar et al, 2010; Orsi et al, 2012; Puri et al, 2013). Irrespective of the exact source of lipids (which remains a matter of debate), forming mammalian phagophores recruit the ULK1 complex and ATG14 (Karanasios et al, 2013), which facilitates the assembly of the autophagy‐specific Class III PI3K complex (Matsunaga et al, 2010). This enables the association of the PI3P‐binding proteins DFCP1 and WIPI2 (Polson et al, 2010), the formation of ATG12‐ATG5:ATG16L1 complexes, and consequent local LC3 lipidation (Dooley et al, 2014). Either mammalian phagophores or omegasomes, or both, stain positively for ULK1, ATG13, ATG101, FIP200, VPS34, BECN1, VPS15, ATG5, ATG12, ATG16L1, DFCP1 as well as for lipidated LC3 family members (Antonioli et al, 2016). In yeast, phagophores are formed at the so‐called “phagophore‐assembly site” or “pre‐autophagosomal structure” (PAS), that is, a site within the cytoplasm enriched in Atg9+ vesicles with a diameter of 30–60 nm that fuse together owing to the tethering activity of Atg1 (the yeast counterpart of ULK1), Atg13, Atg17, Atg19, and Atg31 (Yamamoto et al, 2012; Stanley et al, 2014).
Autophagosomes
Transient, double‐membraned organelles (mean diameter in mammals 0.5–1.5 μm) that mediate cargo sequestration and delivery to lysosomes in the course of macroautophagic responses (Shibutani & Yoshimori, 2014). Autophagosomes originate from, and hence share some biomarker proteins with, closing phagophores (see above). Since autophagosomes are devoid of hydrolytic activity, both ubiquitinated and non‐ubiquitinated autophagy substrates, as well as autophagy receptors, can be detected in this compartment (Klionsky et al, 2016). LC3 is abundant at both the inner and outer membrane of forming autophagosomes. However, it is efficiently removed by Atg4 family members from the surface of closed autophagosomes (Lamb et al, 2013). In the course of functional macroautophagic responses, autophagosomes rapidly fuse with late endosomes or lysosomes (see below) and hence may be difficult to detect as a stable pool. This can be experimentally circumvented by inhibiting fusion or lysosomal acidification (Klionsky et al, 2016).
Amphisomes
Single‐ or double‐membraned organelles that originate from the fusion of autophagosomes and (late) endosomes (Gordon & Seglen, 1988). Amphisomes contain common autophagosomal markers including lipidated LC3, as well as classical endosomal markers like RAB5, RAB7, and RAB11 (the latter of which is also required for autophagosome formation) (Fader et al, 2009; Chandra et al, 2015). Moreover, amphisomes have been proposed to contain small amounts of the lysosomal V‐type ATPase, which would be responsible for progressive acidification of their lumen (Bader et al, 2015).
Autolysosomes
Single‐membraned organelles that form in the course of macroautophagy upon fusion of autophagosomes or amphisomes and lysosomes (Klionsky et al, 2014). Autolysosomes are positive for lysosomal enzymes and classical endo/lysosomal markers, including LAMP1, LAMP2, and the V‐type ATPase, but may display low levels of autophagosomal markers such as lipidated LC3, especially if autophagic flux is high (unless lysosomal hydrolases are pharmacologically or genetically inhibited) (Klionsky et al, 2014). Along similar lines, autophagic substrates and receptors are rapidly degraded within autolysosomes in conditions of elevated autophagic flux, implying that it may be difficult to reveal their presence in this compartment. Once the degradation of autophagy cargos is completed, autolysosomes contribute to the regeneration of the lysosomal pool via ALR (see above) (Yu et al, 2010). Of note, the term autophagolysosome indicates a specific type of autolysosome that forms in the course of some xenophagic responses (Klionsky et al, 2014). In this setting, autophagosomes can engulf entire phagosomes in the absence of membrane fusion, followed by the delivery of a double‐membraned cargo (secluded by the inner autophagosomal membrane plus the phagosomal membrane) to lysosomes (Klionsky et al, 2014). We support the proper semantic and conceptual discrimination between autolysosomes and autophagolysosomes and at the same time discourage the incorrect use of these terms as interchangeable synonyms (which is rather common in the literature).
Concluding remarks
Throughout the past two decades, our understanding of autophagy in mechanistic and pathophysiological terms has progressed tremendously. In parallel, we unveiled a considerable therapeutic potential for molecules that target autophagy and autophagy‐related processes such as LAP. Such a potential remains largely unexploited in the clinic, for reasons that relate to the complex nature of autophagic responses themselves, to the specificity of pharmacological agents developed so far, to the limitations of currently available models, as well as to the imprecise use of autophagy‐related terms. Here, we attempted to provide semantic and conceptual recommendations that may help with this latter issue (Box 1). Our aim is not to provide a rigid vocabulary, but a working framework that can be revised and modified as the field evolves to address the current outstanding questions (Lindqvist et al, 2015). These recommendations are intended to facilitate the dissemination of results and ideas within and outside the field and eventually benefit scientific progress in this and other areas of biological/biomedical investigation.
Box 1:Key recommendations
-
Bona fide autophagic responses deliver cytoplasmic material (of endogenous or exogenous origin) to lysosomes (or vacuoles) for degradation.
-
Microautophagy is a LAMP2A‐independent autophagic response that proceeds upon direct membrane invagination at the surface of the lysosome/vacuole.
-
Endosomal microautophagy is an ESCRT‐dependent, LAMP2A‐independent autophagic response that relies on direct membrane invagination at the surface of late endosomes, occurring either as a bulk process or following HSPA8‐mediated cargo recognition.
-
Chaperone‐mediated autophagy (CMA) is an HSPA8‐ and LAMP2A‐dependent autophagic response that involves the translocation of substrates across the lysosomal membrane.
-
Macroautophagy is a type of autophagic response that relies on the formation of autophagosomes and can be subtyped based upon dependence on specific factors (including—but not limited to—ATG proteins).
-
Selective instances of autophagy should be defined based on the enrichment of a precise substrate, coupled to the requirement of specific molecular factors (such as autophagy receptors).
-
Autophagic flux refers to the global efficacy of autophagic responses, which is generally well represented by the rate at which lysosomes degrade autophagy substrates.
-
Autophagy‐dependent cell death is a form of regulated cell death that can be retarded by pharmacological or genetic inhibition of components of the macroautophagy apparatus.
-
Autosis is a Na+/K+‐ATPase‐mediated type of autophagy‐dependent cell death.
-
Cytoplasm‐to‐vacuole targeting (Cvt), LC3‐associated phagocytosis (LAP), crinophagy, and instances of protein secretion that depend on components of the macroautophagy apparatus are not bona fide autophagic responses.
-
Autophagy substrates are cytoplasmic entities (of endogenous/intracellular or exogenous/extracellular origin) delivered to lysosomal degradation by autophagy.
-
Autophagy receptors are proteins that bind autophagy substrates, allow for their recognition by the autophagy machinery, and get degraded within lysosomes in the course of functional autophagic responses.
-
Autophagy adaptors are proteins that interact with Atg8 family members, hence conferring additional functions to the autophagosome, but are not involved in cargo recognition.
-
Phagophores (also called isolation membranes) are the precursors of autophagosomes.
-
Autophagosomes are transient, double‐membraned organelles that mediate cargo sequestration and delivery to lysosomes in the course of macroautophagic responses.
-
Amphisomes are single‐ or double‐membraned organelles that originate from the fusion of autophagosomes and (late) endosomes.
-
Autolysosomes are single‐membraned organelles that form in the course of macroautophagy upon fusion of autophagosomes or amphisomes with lysosomes.
-
Autophagolysosomes are a specific type of autolysosome that forms in the course of xenophagic responses targeting intact or ruptured phagosomes.
References
Agarraberes FA, Terlecky SR, Dice JF (1997) An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol 137: 825–834
Al Rawi S, Louvet‐Vallee S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V (2011) Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 334: 1144–1147
Amaravadi R, Kimmelman AC, White E (2016) Recent insights into the function of autophagy in cancer. Genes Dev 30: 1913–1930
Amaya C, Fader CM, Colombo MI (2015) Autophagy and proteins involved in vesicular trafficking. FEBS Lett 589: 3343–3353
Antonioli M, Di Rienzo M, Piacentini M, Fimia GM (2016) Emerging mechanisms in initiating and terminating autophagy. Trends Biochem Sci 42: 28–41
Arias E, Koga H, Diaz A, Mocholi E, Patel B, Cuervo AM (2015) Lysosomal mTORC2/PHLPP1/Akt regulate chaperone‐mediated autophagy. Mol Cell 59: 270–284
Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, Hesse M, Furst DO, Saftig P, Saint R, Fleischmann BK, Hoch M, Hohfeld J (2010) Chaperone‐assisted selective autophagy is essential for muscle maintenance. Curr Biol 20: 143–148
Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT (2008) Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3‐phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182: 685–701
Bader CA, Shandala T, Ng YS, Johnson IR, Brooks DA (2015) Atg9 is required for intraluminal vesicles in amphisomes and autolysosomes. Biol Open 4: 1345–1355
Bakowski MA, Desjardins CA, Smelkinson MG, Dunbar TL, Lopez‐Moyado IF, Rifkin SA, Cuomo CA, Troemel ER (2014) Ubiquitin‐mediated response to microsporidia and virus infection in Caenorhabditis elegans. PLoS Pathog 10: e1004200
Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM (2008) The chaperone‐mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol 28: 5747–5763
Bandyopadhyay U, Sridhar S, Kaushik S, Kiffin R, Cuervo AM (2010) Identification of regulators of chaperone‐mediated autophagy. Mol Cell 39: 535–547
Berry DL, Baehrecke EH (2007) Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 131: 1137–1148
Bertipaglia C, Schneider S, Jakobi AJ, Tarafder AK, Bykov YS, Picco A, Kukulski W, Kosinski J, Hagen WJ, Ravichandran AC, Wilmanns M, Kaksonen M, Briggs JA, Sachse C (2016) Higher‐order assemblies of oligomeric cargo receptor complexes form the membrane scaffold of the Cvt vesicle. EMBO Rep 17: 1044–1060
Bhattacharyya S, Yu H, Mim C, Matouschek A (2014) Regulated protein turnover: snapshots of the proteasome in action. Nat Rev Mol Cell Biol 15: 122–133
Birgisdottir AB, Lamark T, Johansen T (2013) The LIR motif ‐ crucial for selective autophagy. J Cell Sci 126: 3237–3247
Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. J Cell Biol 171: 603–614
Boya P, Gonzalez‐Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G (2005) Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 25: 1025–1040
Boya P, Reggiori F, Codogno P (2013) Emerging regulation and functions of autophagy. Nat Cell Biol 15: 713–720
Buchan JR, Kolaitis RM, Taylor JP, Parker R (2013) Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153: 1461–1474
Burnett SF, Farre JC, Nazarko TY, Subramani S (2015) Peroxisomal Pex3 activates selective autophagy of peroxisomes via interaction with the pexophagy receptor Atg30. J Biol Chem 290: 8623–8631
Chandra P, Ghanwat S, Matta SK, Yadav SS, Mehta M, Siddiqui Z, Singh A, Kumar D (2015) Mycobacterium tuberculosis inhibits RAB7 recruitment to selectively modulate autophagy flux in macrophages. Sci Rep 5: 16320
Chang TK, Shravage BV, Hayes SD, Powers CM, Simin RT, Wade Harper J, Baehrecke EH (2013) Uba1 functions in Atg7‐ and Atg3‐independent autophagy. Nat Cell Biol 15: 1067–1078
Chanoca A, Kovinich N, Burkel B, Stecha S, Bohorquez‐Restrepo A, Ueda T, Eliceiri KW, Grotewold E, Otegui MS (2015) Anthocyanin vacuolar inclusions form by a microautophagy mechanism. Plant Cell 27: 2545–2559
Chauhan S, Mandell MA, Deretic V (2015) IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol Cell 58: 507–521
Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T, Choi SW, Peters R, Mandell M, Bruun JA, Johansen T, Deretic V (2016) TRIMs and galectins globally cooperate and TRIM16 and galectin‐3 co‐direct autophagy in endomembrane damage homeostasis. Dev Cell 39: 13–27
Chen D, Fan W, Lu Y, Ding X, Chen S, Zhong Q (2012) A mammalian autophagosome maturation mechanism mediated by TECPR1 and the Atg12‐Atg5 conjugate. Mol Cell 45: 629–641
Cherra SJ III, Dagda RK, Chu CT (2010a) Review: autophagy and neurodegeneration: survival at a cost? Neuropathol Appl Neurobiol 36: 125–132
Cherra SJ III, Kulich SM, Uechi G, Balasubramani M, Mountzouris J, Day BW, Chu CT (2010b) Regulation of the autophagy protein LC3 by phosphorylation. J Cell Biol 190: 533–539
Chiang HL, Terlecky SR, Plant CP, Dice JF (1989) A role for a 70‐kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246: 382–385
Choi AM, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368: 651–662
Choi J, Park S, Biering SB, Selleck E, Liu CY, Zhang X, Fujita N, Saitoh T, Akira S, Yoshimori T, Sibley LD, Hwang S, Virgin HW (2014) The parasitophorous vacuole membrane of Toxoplasma gondii is targeted for disruption by ubiquitin‐like conjugation systems of autophagy. Immunity 40: 924–935
Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, Sachleben JR, Asara JM, Locasale JW, Karczmar GS, Macleod KF (2015) Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep 16: 1145–1163
Chu CT (2011) Autophagy in different flavors: dysregulated protein degradation in neurological diseases. Neurobiol Dis 43: 1–3
Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, Qiang Wang KZ, Zhu J, Klein‐Seetharaman J, Balasubramanian K, Amoscato AA, Borisenko G, Huang Z, Gusdon AM, Cheikhi A, Steer EK et al (2013) Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 15: 1197–1205
Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441: 1162–1166
Codogno P, Mehrpour M, Proikas‐Cezanne T (2012) Canonical and non‐canonical autophagy: variations on a common theme of self‐eating? Nat Rev Mol Cell Biol 13: 7–12
Cohen‐Kaplan V, Livneh I, Avni N, Fabre B, Ziv T, Kwon YT, Ciechanover A (2016) p62‐ and ubiquitin‐dependent stress‐induced autophagy of the mammalian 26S proteasome. Proc Natl Acad Sci USA 113: E7490–E7499
Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A (2010) NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 16: 90–97
Cuervo AM, Palmer A, Rivett AJ, Knecht E (1995) Degradation of proteasomes by lysosomes in rat liver. Eur J Biochem 227: 792–800
Cuervo AM, Dice JF (1996) A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273: 501–503
Cuervo AM, Dice JF (2000) Age‐related decline in chaperone‐mediated autophagy. J Biol Chem 275: 31505–31513
Cuervo AM, Wong E (2014) Chaperone‐mediated autophagy: roles in disease and aging. Cell Res 24: 92–104
D'Eletto M, Farrace MG, Rossin F, Strappazzon F, Giacomo GD, Cecconi F, Melino G, Sepe S, Moreno S, Fimia GM, Falasca L, Nardacci R, Piacentini M (2012) Type 2 transglutaminase is involved in the autophagy‐dependent clearance of ubiquitinated proteins. Cell Death Differ 19: 1228–1238
Demontis F, Perrimon N (2010) FOXO/4E‐BP signaling in Drosophila muscles regulates organism‐wide proteostasis during aging. Cell 143: 813–825
Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, Kumar S (2009) Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol 19: 1741–1746
Denton D, Aung‐Htut MT, Lorensuhewa N, Nicolson S, Zhu W, Mills K, Cakouros D, Bergmann A, Kumar S (2013) UTX coordinates steroid hormone‐mediated autophagy and cell death. Nat Commun 4: 2916
Denton D, Xu T, Kumar S (2015) Autophagy as a pro‐death pathway. Immunol Cell Biol 93: 35–42
Deosaran E, Larsen KB, Hua R, Sargent G, Wang Y, Kim S, Lamark T, Jauregui M, Law K, Lippincott‐Schwartz J, Brech A, Johansen T, Kim PK (2013) NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci 126: 939–952
Deretic V, Saitoh T, Akira S (2013) Autophagy in infection, inflammation and immunity. Nat Rev Immunol 13: 722–737
Diao J, Liu R, Rong Y, Zhao M, Zhang J, Lai Y, Zhou Q, Wilz LM, Li J, Vivona S, Pfuetzner RA, Brunger AT, Zhong Q (2015) ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 520: 563–566
Dice JF (1990) Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci 15: 305–309
Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA (2014) WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12‐5‐16L1. Mol Cell 55: 238–252
Dou Z, Xu C, Donahue G, Shimi T, Pan JA, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, Catanzaro JM, Ricketts MD, Lamark T, Adam SA, Marmorstein R, Zong WX, Johansen T, Goldman RD, Adams PD, Berger SL (2015) Autophagy mediates degradation of nuclear lamina. Nature 527: 105–109
Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, Menon S, Wang Z, Honda A, Pardee G, Cantwell J, Luu C, Cornella‐Taracido I, Harrington E, Fekkes P, Lei H, Fang Q, Digan ME, Burdick D, Powers AF et al (2014) Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol 16: 1069–1079
Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V (2011) Autophagy‐based unconventional secretory pathway for extracellular delivery of IL‐1beta. EMBO J 30: 4701–4711
Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ (2011) Phosphorylation of ULK1 (hATG1) by AMP‐activated protein kinase connects energy sensing to mitophagy. Science 331: 456–461
Eng CH, Wang Z, Tkach D, Toral‐Barza L, Ugwonali S, Liu S, Fitzgerald SL, George E, Frias E, Cochran N, De Jesus R, McAllister G, Hoffman GR, Bray K, Lemon L, Lucas J, Fantin VR, Abraham RT, Murphy LO, Nyfeler B (2016) Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci USA 113: 182–187
Eskelinen EL, Cuervo AM, Taylor MR, Nishino I, Blum JS, Dice JF, Sandoval IV, Lippincott‐Schwartz J, August JT, Saftig P (2005) Unifying nomenclature for the isoforms of the lysosomal membrane protein LAMP‐2. Traffic 6: 1058–1061
Eskelinen EL, Reggiori F, Baba M, Kovacs AL, Seglen PO (2011) Seeing is believing: the impact of electron microscopy on autophagy research. Autophagy 7: 935–956
Fader CM, Sanchez DG, Mestre MB, Colombo MI (2009) TI‐VAMP/VAMP7 and VAMP3/cellubrevin: two v‐SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim Biophys Acta 1793: 1901–1916
Fader CM, Salassa BN, Grosso RA, Vergara AN, Colombo MI (2016) Hemin induces mitophagy in a leukemic erythroblast cell line. Biol Cell 108: 77–95
Fan W, Nassiri A, Zhong Q (2011) Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L). Proc Natl Acad Sci USA 108: 7769–7774
Farre JC, Subramani S (2004) Peroxisome turnover by micropexophagy: an autophagy‐related process. Trends Cell Biol 14: 515–523
Farre JC, Manjithaya R, Mathewson RD, Subramani S (2008) PpAtg30 tags peroxisomes for turnover by selective autophagy. Dev Cell 14: 365–376
Farre JC, Subramani S (2016) Mechanistic insights into selective autophagy pathways: lessons from yeast. Nat Rev Mol Cell Biol 17: 537–552
Filimonenko M, Isakson P, Finley KD, Anderson M, Jeong H, Melia TJ, Bartlett BJ, Myers KM, Birkeland HC, Lamark T, Krainc D, Brech A, Stenmark H, Simonsen A, Yamamoto A (2010) The selective macroautophagic degradation of aggregated proteins requires the PI3P‐binding protein Alfy. Mol Cell 38: 265–279
Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F (2007) Ambra1 regulates autophagy and development of the nervous system. Nature 447: 1121–1125
Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M (2011) Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol 13: 1335–1343
Folick A, Oakley HD, Yu Y, Armstrong EH, Kumari M, Sanor L, Moore DD, Ortlund EA, Zechner R, Wang MC (2015) Aging. Lysosomal signaling molecules regulate longevity in Caenorhabditis elegans. Science 347: 83–86
Foot N, Henshall T, Kumar S (2017) Ubiquitination and the regulation of membrane proteins. Physiol Rev 97: 253–281
Fracchiolla D, Sawa‐Makarska J, Zens B, Ruiter A, Zaffagnini G, Brezovich A, Romanov J, Runggatscher K, Kraft C, Zagrovic B, Martens S (2016) Mechanism of cargo‐directed Atg8 conjugation during selective autophagy. Elife 5: e18544
Fumagalli F, Noack J, Bergmann TJ, Cebollero E, Pisoni GB, Fasana E, Fregno I, Galli C, Loi M, Solda T, D'Antuono R, Raimondi A, Jung M, Melnyk A, Schorr S, Schreiber A, Simonelli L, Varani L, Wilson‐Zbinden C, Zerbe O et al (2016) Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat Cell Biol 18: 1173–1184
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El‐Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W et al (2012) Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 19: 107–120
Galluzzi L, Pietrocola F, Levine B, Kroemer G (2014) Metabolic control of autophagy. Cell 159: 1263–1276
Galluzzi L, Bravo‐San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico‐Petruzzelli M, Baehrecke EH, Bazan NG, Bertrand MJ, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Campanella M et al (2015a) Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ 22: 58–73
Galluzzi L, Pietrocola F, Bravo‐San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, Kimmelman A, Kumar S, Levine B, Maiuri MC, Martin SJ, Penninger J, Piacentini M, Rubinsztein DC, Simon HU, Simonsen A et al (2015b) Autophagy in malignant transformation and cancer progression. EMBO J 34: 856–880
Galluzzi L, Bravo‐San Pedro JM, Blomgren K, Kroemer G (2016) Autophagy in acute brain injury. Nat Rev Neurosci 17: 467–484
Galluzzi L, Bravo‐San Pedro JM, Demaria S, Formenti SC, Kroemer G (2017a) Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat Rev Clin Oncol 14: 247–258
Galluzzi L, Bravo‐San Pedro JM, Levine B, Green DR, Kroemer G (2017b) Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov https://doi.org/10.1038/nrd.2017.22.
Gan B, Peng X, Nagy T, Alcaraz A, Gu H, Guan JL (2006) Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC‐mTOR signaling pathways. J Cell Biol 175: 121–133
Gannage M, Dormann D, Albrecht R, Dengjel J, Torossi T, Ramer PC, Lee M, Strowig T, Arrey F, Conenello G, Pypaert M, Andersen J, Garcia‐Sastre A, Munz C (2009) Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6: 367–380
Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010) PINK1/Parkin‐mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12: 119–131
Gerstenmaier L, Pilla R, Herrmann L, Herrmann H, Prado M, Villafano GJ, Kolonko M, Reimer R, Soldati T, King JS, Hagedorn M (2015) The autophagic machinery ensures nonlytic transmission of mycobacteria. Proc Natl Acad Sci USA 112: E687–E692
Gomes LC, Di Benedetto G, Scorrano L (2011a) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13: 589–598
Gomes LC, Di Benedetto G, Scorrano L (2011b) Essential amino acids and glutamine regulate induction of mitochondrial elongation during autophagy. Cell Cycle 10: 2635–2639
Gomez‐Sanchez JA, Carty L, Iruarrizaga‐Lejarreta M, Palomo‐Irigoyen M, Varela‐Rey M, Griffith M, Hantke J, Macias‐Camara N, Azkargorta M, Aurrekoetxea I, De Juan VG, Jefferies HB, Aspichueta P, Elortza F, Aransay AM, Martinez‐Chantar ML, Baas F, Mato JM, Mirsky R, Woodhoo A et al (2015) Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J Cell Biol 210: 153–168
Gong L, Cullinane M, Treerat P, Ramm G, Prescott M, Adler B, Boyce JD, Devenish RJ (2011) The Burkholderia pseudomallei type III secretion system and BopA are required for evasion of LC3‐associated phagocytosis. PLoS ONE 6: e17852
Gonzalez‐Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquere S, Eskelinen EL, Pierron G, Saftig P, Kroemer G (2005) The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci 118: 3091–3102
Gordon PB, Seglen PO (1988) Prelysosomal convergence of autophagic and endocytic pathways. Biochem Biophys Res Commun 151: 40–47
Grasso D, Ropolo A, Lo Re A, Boggio V, Molejon MI, Iovanna JL, Gonzalez CD, Urrutia R, Vaccaro MI (2011) Zymophagy, a novel selective autophagy pathway mediated by VMP1‐USP9x‐p62, prevents pancreatic cell death. J Biol Chem 286: 8308–8324
Green DR, Levine B (2014) To be or not to be? How selective autophagy and cell death govern cell fate. Cell 157: 65–75
Griffiths RE, Kupzig S, Cogan N, Mankelow TJ, Betin VM, Trakarnsanga K, Massey EJ, Lane JD, Parsons SF, Anstee DJ (2012) Maturing reticulocytes internalize plasma membrane in glycophorin A‐containing vesicles that fuse with autophagosomes before exocytosis. Blood 119: 6296–6306
Gutierrez MG, Munafo DB, Beron W, Colombo MI (2004) Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci 117: 2687–2697
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki‐Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: 885–889
Hasegawa J, Iwamoto R, Otomo T, Nezu A, Hamasaki M, Yoshimori T (2016) Autophagosome‐lysosome fusion in neurons requires INPP5E, a protein associated with Joubert syndrome. EMBO J 35: 1853–1867
Heath RJ, Goel G, Baxt LA, Rush JS, Mohanan V, Paulus GL, Jani V, Lassen KG, Xavier RJ (2016) RNF166 determines recruitment of adaptor proteins during antibacterial autophagy. Cell Rep 17: 2183–2194
Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW (2015) The PINK1‐PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 60: 7–20
Hjerpe R, Bett JS, Keuss MJ, Solovyova A, McWilliams TG, Johnson C, Sahu I, Varghese J, Wood N, Wightman M, Osborne G, Bates GP, Glickman MH, Trost M, Knebel A, Marchesi F, Kurz T (2016) UBQLN2 mediates autophagy‐independent protein aggregate clearance by the proteasome. Cell 166: 935–949
Huang J, Brumell JH (2014) Bacteria‐autophagy interplay: a battle for survival. Nat Rev Microbiol 12: 101–114
Huang H, Kawamata T, Horie T, Tsugawa H, Nakayama Y, Ohsumi Y, Fukusaki E (2015) Bulk RNA degradation by nitrogen starvation‐induced autophagy in yeast. EMBO J 34: 154–168
Huang K, Liu D (2016) Targeting non‐canonical autophagy overcomes erlotinib resistance in tongue cancer. Tumour Biol 37: 9625–9633
Hung YH, Chen LM, Yang JY, Yang WY (2013) Spatiotemporally controlled induction of autophagy‐mediated lysosome turnover. Nat Commun 4: 2111
Hwang S, Maloney NS, Bruinsma MW, Goel G, Duan E, Zhang L, Shrestha B, Diamond MS, Dani A, Sosnovtsev SV, Green KY, Lopez‐Otin C, Xavier RJ, Thackray LB, Virgin HW (2012) Nondegradative role of Atg5‐Atg12/Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe 11: 397–409
Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, Noda T, Ohsumi Y (2000) A ubiquitin‐like system mediates protein lipidation. Nature 408: 488–492
Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648–657
Itakura E, Kishi C, Inoue K, Mizushima N (2008) Beclin 1 forms two distinct phosphatidylinositol 3‐kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 19: 5360–5372
Itakura E, Kishi‐Itakura C, Mizushima N (2012) The hairpin‐type tail‐anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151: 1256–1269
Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL (2004) Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 117: 4837–4848
Jia K, Hart AC, Levine B (2007) Autophagy genes protect against disease caused by polyglutamine expansion proteins in Caenorhabditis elegans. Autophagy 3: 21–25
Jia K, Thomas C, Akbar M, Sun Q, Adams‐Huet B, Gilpin C, Levine B (2009) Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling‐regulated pathogen resistance. Proc Natl Acad Sci USA 106: 14564–14569
Joachim J, Jefferies HB, Razi M, Frith D, Snijders AP, Chakravarty P, Judith D, Tooze SA (2015) Activation of ULK kinase and autophagy by GABARAP trafficking from the centrosome is regulated by WAC and GM130. Mol Cell 60: 899–913
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH (2009) ULK‐Atg13‐FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20: 1992–2003
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728
Kagan VE, Jiang J, Huang Z, Tyurina YY, Desbourdes C, Cottet‐Rousselle C, Dar HH, Verma M, Tyurin VA, Kapralov AA, Cheikhi A, Mao G, Stolz D, St Croix CM, Watkins S, Shen Z, Li Y, Greenberg ML, Tokarska‐Schlattner M, Boissan M et al (2016) NDPK‐D (NM23‐H4)‐mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Differ 23: 1140–1151
Kaizuka T, Morishita H, Hama Y, Tsukamoto S, Matsui T, Toyota Y, Kodama A, Ishihara T, Mizushima T, Mizushima N (2016) An autophagic flux probe that releases an internal control. Mol Cell 64: 835–849
Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ (2009) Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 17: 98–109
Karanasios E, Stapleton E, Manifava M, Kaizuka T, Mizushima N, Walker SA, Ktistakis NT (2013) Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J Cell Sci 126: 5224–5238
Karanasios E, Walker SA, Okkenhaug H, Manifava M, Hummel E, Zimmermann H, Ahmed Q, Domart MC, Collinson L, Ktistakis NT (2016) Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat Commun 7: 12420
Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A (2011) A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol 18: 1042–1052
Kaur J, Debnath J (2015) Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol 16: 461–472
Kaushik S, Massey AC, Cuervo AM (2006) Lysosome membrane lipid microdomains: novel regulators of chaperone‐mediated autophagy. EMBO J 25: 3921–3933
Kaushik S, Cuervo AM (2012) Chaperone‐mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 22: 407–417
Kaushik S, Cuervo AM (2015) Degradation of lipid droplet‐associated proteins by chaperone‐mediated autophagy facilitates lipolysis. Nat Cell Biol 17: 759–770
Kaushik S, Cuervo AM (2016) AMPK‐dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 12: 432–438
Kawabata T, Yoshimori T (2016) Beyond starvation: an update on the autophagic machinery and its functions. J Mol Cell Cardiol 95: 2–10
Kelly SP, Bedwell DM (2015) Both the autophagy and proteasomal pathways facilitate the Ubp3p‐dependent depletion of a subset of translation and RNA turnover factors during nitrogen starvation in Saccharomyces cerevisiae. RNA 21: 898–910
Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, Mauthe M, Katona I, Qualmann B, Weis J, Reggiori F, Kurth I, Hubner CA, Dikic I (2015) Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522: 354–358
Khaminets A, Behl C, Dikic I (2016) Ubiquitin‐dependent and independent signals in selective autophagy. Trends Cell Biol 26: 6–16
Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T (2001a) Beclin‐phosphatidylinositol 3‐kinase complex functions at the trans‐Golgi network. EMBO Rep 2: 330–335
Kihara A, Noda T, Ishihara N, Ohsumi Y (2001b) Two distinct Vps34 phosphatidylinositol 3‐kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol 152: 519–530
Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141
Kim JJ, Lee HM, Shin DM, Kim W, Yuk JM, Jin HS, Lee SH, Cha GH, Kim JM, Lee ZW, Shin SJ, Yoo H, Park YK, Park JB, Chung J, Yoshimori T, Jo EK (2012) Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe 11: 457–468
Kim BW, Hong SB, Kim JH, Kwon DH, Song HK (2013a) Structural basis for recognition of autophagic receptor NDP52 by the sugar receptor galectin‐8. Nat Commun 4: 1613
Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL (2013b) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152: 290–303
Kim KH, Lee MS (2014) Autophagy–a key player in cellular and body metabolism. Nat Rev Endocrinol 10: 322–337
Kimura T, Jain A, Choi SW, Mandell MA, Schroder K, Johansen T, Deretic V (2015) TRIM‐mediated precision autophagy targets cytoplasmic regulators of innate immunity. J Cell Biol 210: 973–989
Kimura T, Mandell M, Deretic V (2016) Precision autophagy directed by receptor regulators – emerging examples within the TRIM family. J Cell Sci 129: 881–891
Kimura T, Jia J, Kumar S, Choi SW, Gu Y, Mudd M, Dupont N, Jiang S, Peters R, Farzam F, Jain A, Lidke KA, Adams CM, Johansen T, Deretic V (2017) Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J 36: 42–60
Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Overvatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T (2009a) A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 33: 505–516
Kirkin V, McEwan DG, Novak I, Dikic I (2009b) A role for ubiquitin in selective autophagy. Mol Cell 34: 259–269
Kissova I, Salin B, Schaeffer J, Bhatia S, Manon S, Camougrand N (2007) Selective and non‐selective autophagic degradation of mitochondria in yeast. Autophagy 3: 329–336
Klionsky DJ, Eskelinen EL, Deretic V (2014) Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes.. wait I'm confused. Autophagy 10: 549–551
Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, Agarwal R, Aghi MK, Agnello M, Agostinis P, Aguilar PV, Aguirre‐Ghiso J, Airoldi EM et al (2016) Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12: 1–222
Knaevelsrud H, Soreng K, Raiborg C, Haberg K, Rasmuson F, Brech A, Liestol K, Rusten TE, Stenmark H, Neufeld TP, Carlsson SR, Simonsen A (2013) Membrane remodeling by the PX‐BAR protein SNX18 promotes autophagosome formation. J Cell Biol 202: 331–349
Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T (2005) Impairment of starvation‐induced and constitutive autophagy in Atg7‐deficient mice. J Cell Biol 169: 425–434
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441: 880–884
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A et al (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy‐deficient mice. Cell 131: 1149–1163
Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, Cuervo AM (2011) Chaperone‐mediated autophagy is required for tumor growth. Sci Transl Med 3: 109ra117
Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, Behl C, Terzic J, Dikic I (2013) Ubiquitin‐independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci 126: 580–592
Kraft C, Deplazes A, Sohrmann M, Peter M (2008) Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol 10: 602–610
Krick R, Muehe Y, Prick T, Bremer S, Schlotterhose P, Eskelinen EL, Millen J, Goldfarb DS, Thumm M (2008) Piecemeal microautophagy of the nucleus requires the core macroautophagy genes. Mol Biol Cell 19: 4492–4505
Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El‐Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nunez G, Peter ME, Tschopp J, Yuan J, Piacentini M et al (2009) Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 16: 3–11
Ktistakis NT, Tooze SA (2016) Digesting the expanding mechanisms of autophagy. Trends Cell Biol 26: 624–635
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N (2004) The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036
Kuma A, Matsui M, Mizushima N (2007) LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 3: 323–328
Kvam E, Goldfarb DS (2004) Nvj1p is the outer‐nuclear‐membrane receptor for oxysterol‐binding protein homolog Osh1p in Saccharomyces cerevisiae. J Cell Sci 117: 4959–4968
Kvam E, Goldfarb DS (2007) Nucleus‐vacuole junctions and piecemeal microautophagy of the nucleus in Saccharomyces cerevisiae. Autophagy 3: 85–92
Lam GY, Cemma M, Muise AM, Higgins DE, Brumell JH (2013) Host and bacterial factors that regulate LC3 recruitment to Listeria monocytogenes during the early stages of macrophage infection. Autophagy 9: 985–995
Lamb CA, Yoshimori T, Tooze SA (2013) The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 14: 759–774
Lapierre LR, Gelino S, Melendez A, Hansen M (2011) Autophagy and lipid metabolism coordinately modulate life span in germline‐less Caenorhabditis elegans. Curr Biol 21: 1507–1514
Lapierre LR, Silvestrini MJ, Nunez L, Ames K, Wong S, Le TT, Hansen M, Melendez A (2013) Autophagy genes are required for normal lipid levels in Caenorhabditis elegans. Autophagy 9: 278–286
Lapierre LR, Kumsta C, Sandri M, Ballabio A, Hansen M (2015) Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 11: 867–880
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314
Lee JW, Park S, Takahashi Y, Wang HG (2010) The association of AMPK with ULK1 regulates autophagy. PLoS ONE 5: e15394
Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, Moore DD (2014) Nutrient‐sensing nuclear receptors coordinate autophagy. Nature 516: 112–115
Lee MY, Sumpter Jr R, Zou Z, Sirasanagandla S, Wei Y, Mishra P, Rosewich H, Crane DI, Levine B (2017) Peroxisomal protein PEX13 functions in selective autophagy. EMBO Rep 18: 48–60
Levine B, Mizushima N, Virgin HW (2011) Autophagy in immunity and inflammation. Nature 469: 323–335
Li WW, Li J, Bao JK (2012) Microautophagy: lesser‐known self‐eating. Cell Mol Life Sci 69: 1125–1136
Li S, Wandel MP, Li F, Liu Z, He C, Wu J, Shi Y, Randow F (2013) Sterical hindrance promotes selectivity of the autophagy cargo receptor NDP52 for the danger receptor galectin‐8 in antibacterial autophagy. Sci Signal 6: ra9
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402: 672–676
Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, Jung JU (2008) Beclin1‐binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 10: 776–787
Lim J, Yue Z (2015) Neuronal aggregates: formation, clearance, and spreading. Dev Cell 32: 491–501
Lindqvist LM, Simon AK, Baehrecke EH (2015) Current questions and possible controversies in autophagy. Cell Death Discov 1: 15036
Lipatova Z, Shah AH, Kim JJ, Mulholland JW, Segev N (2013) Regulation of ER‐phagy by a Ypt/Rab GTPase module. Mol Biol Cell 24: 3133–3144
Liu Y, Schiff M, Czymmek K, Talloczy Z, Levine B, Dinesh‐Kumar SP (2005) Autophagy regulates programmed cell death during the plant innate immune response. Cell 121: 567–577
Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S et al (2012) Mitochondrial outer‐membrane protein FUNDC1 mediates hypoxia‐induced mitophagy in mammalian cells. Nat Cell Biol 14: 177–185
Liu Y, Shoji‐Kawata S, Sumpter RM Jr, Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, Shaw SY, Clarke PG, Puyal J, Levine B (2013) Autosis is a Na+, K+‐ATPase‐regulated form of cell death triggered by autophagy‐inducing peptides, starvation, and hypoxia‐ischemia. Proc Natl Acad Sci USA 110: 20364–20371
Liu EY, Xu N, O'Prey J, Lao LY, Joshi S, Long JS, O'Prey M, Croft DR, Beaumatin F, Baudot AD, Mrschtik M, Rosenfeldt M, Zhang Y, Gillespie DA, Ryan KM (2015a) Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc Natl Acad Sci USA 112: 773–778
Liu XM, Sun LL, Hu W, Ding YH, Dong MQ, Du LL (2015b) ESCRTs cooperate with a selective autophagy receptor to mediate vacuolar targeting of soluble cargos. Mol Cell 59: 1035–1042
Lock R, Kenific CM, Leidal AM, Salas E, Debnath J (2014) Autophagy‐dependent production of secreted factors facilitates oncogenic RAS‐driven invasion. Cancer Discov 4: 466–479
Loos B, du Toit A, Hofmeyr JH (2014) Defining and measuring autophagosome flux‐concept and reality. Autophagy 10: 2087–2096
Lopez‐Otin C, Galluzzi L, Freije JM, Madeo F, Kroemer G (2016) Metabolic control of longevity. Cell 166: 802–821
Lu Q, Wu F, Zhang H (2013) Aggrephagy: lessons from Caenorhabditis elegans. Biochem J 452: 381–390
Lu J, He L, Behrends C, Araki M, Araki K, Jun Wang Q, Catanzaro JM, Friedman SL, Zong WX, Fiel MI, Li M, Yue Z (2014a) NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L‐linked phosphatidylinositol‐3 kinase III activity. Nat Commun 5: 3920
Lu K, Psakhye I, Jentsch S (2014b) Autophagic clearance of polyQ proteins mediated by ubiquitin‐Atg8 adaptors of the conserved CUET protein family. Cell 158: 549–563
Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, Yoshimori T (2013) Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 32: 2336–2347
Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, Quaegebeur A, Schoors S, Georgiadou M, Wouters J, Vinckier S, Vankelecom H, Garmyn M, Vion AC, Radtke F, Boulanger C, Gerhardt H, Dejana E, Dewerchin M, Ghesquiere B et al (2014) Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 26: 190–206
Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC (2014) Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509: 105–109
Mandell MA, Jain A, Arko‐Mensah J, Chauhan S, Kimura T, Dinkins C, Silvestri G, Munch J, Kirchhoff F, Simonsen A, Wei Y, Levine B, Johansen T, Deretic V (2014) TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell 30: 394–409
Manjithaya R, Anjard C, Loomis WF, Subramani S (2010) Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol 188: 537–546
Marino G, Fernandez AF, Cabrera S, Lundberg YW, Cabanillas R, Rodriguez F, Salvador‐Montoliu N, Vega JA, Germana A, Fueyo A, Freije JM, Lopez‐Otin C (2010) Autophagy is essential for mouse sense of balance. J Clin Invest 120: 2331–2344
Marshall RS, Li F, Gemperline DC, Book AJ, Vierstra RD (2015) Autophagic degradation of the 26S proteasome is mediated by the dual ATG8/ubiquitin receptor RPN10 in Arabidopsis. Mol Cell 58: 1053–1066
Marshall RS, McLoughlin F, Vierstra RD (2016) Autophagic turnover of inactive 26S proteasomes in yeast is directed by the ubiquitin receptor Cue5 and the Hsp42 chaperone. Cell Rep 16: 1717–1732
Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan JL, Tan H, Peng J, Kanneganti TD, Virgin HW, Green DR (2015) Molecular characterization of LC3‐associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol 17: 893–906
Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, Li QZ, Yan M, Janke L, Guy C, Linkermann A, Virgin HW, Green DR (2016) Noncanonical autophagy inhibits the autoinflammatory, lupus‐like response to dying cells. Nature 533: 115–119
Marzella L, Ahlberg J, Glaumann H (1981) Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Arch B Cell Pathol Incl Mol Pathol 36: 219–234
Masini M, Lupi R, Bugliani M, Boggi U, Filipponi F, Masiello P, Marchetti P (2009) A role for autophagy in beta‐cell life and death. Islets 1: 157–159
Maskey D, Yousefi S, Schmid I, Zlobec I, Perren A, Friis R, Simon HU (2013) ATG5 is induced by DNA‐damaging agents and promotes mitotic catastrophe independent of autophagy. Nat Commun 4: 2130
Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM (2006) Consequences of the selective blockage of chaperone‐mediated autophagy. Proc Natl Acad Sci USA 103: 5805–5810
Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama‐Noda K, Ichimura T, Isobe T, Akira S, Noda T, Yoshimori T (2009) Two Beclin 1‐binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11: 385–396
Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, Noda T, Yoshimori T (2010) Autophagy requires endoplasmic reticulum targeting of the PI3‐kinase complex via Atg14L. J Cell Biol 190: 511–521
Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A (2012) Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 8: 200–212
McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K, Kirchof A, Gatti E, Helfrich MH, Wakatsuki S, Behrends C, Pierre P, Dikic I (2015) PLEKHM1 regulates autophagosome‐lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell 57: 39–54
Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B (2003) Autophagy genes are essential for dauer development and life‐span extension in Caenorhabditis elegans. Science 301: 1387–1391
Menzies FM, Fleming A, Rubinsztein DC (2015) Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci 16: 345–357
Mijaljica D, Prescott M, Devenish RJ (2012) A late form of nucleophagy in Saccharomyces cerevisiae. PLoS ONE 7: e40013
Mindell JA (2012) Lysosomal acidification mechanisms. Annu Rev Physiol 74: 69–86
Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y (1998) A protein conjugation system essential for autophagy. Nature 395: 395–398
Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, Nakatogawa H (2015) Receptor‐mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 522: 359–362
Moore AS, Holzbaur EL (2016) Dynamic recruitment and activation of ALS‐associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci USA 113: E3349–E3358
Morozova K, Clement CC, Kaushik S, Stiller B, Arias E, Ahmad A, Rauch JN, Chatterjee V, Melis C, Scharf B, Gestwicki JE, Cuervo AM, Zuiderweg ER, Santambrogio L (2016) Structural and biological interaction of hsc‐70 protein with phosphatidylserine in endosomal microautophagy. J Biol Chem 291: 18096–18106
Mortensen M, Ferguson DJ, Edelmann M, Kessler B, Morten KJ, Komatsu M, Simon AK (2010) Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci USA 107: 832–837
Moscat J, Karin M, Diaz‐Meco MT (2016) p62 in cancer: signaling adaptor beyond autophagy. Cell 167: 606–609
Motley AM, Nuttall JM, Hettema EH (2012) Pex3‐anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. EMBO J 31: 2852–2868
Moy RH, Gold B, Molleston JM, Schad V, Yanger K, Salzano MV, Yagi Y, Fitzgerald KA, Stanger BZ, Soldan SS, Cherry S (2014) Antiviral autophagy restricts Rift Valley fever virus infection and is conserved from flies to mammals. Immunity 40: 51–65
Mrschtik M, O'Prey J, Lao LY, Long JS, Beaumatin F, Strachan D, O'Prey M, Skommer J, Ryan KM (2015) DRAM‐3 modulates autophagy and promotes cell survival in the absence of glucose. Cell Death Differ 22: 1714–1726
von Muhlinen N, Akutsu M, Ravenhill BJ, Foeglein A, Bloor S, Rutherford TJ, Freund SM, Komander D, Randow F (2012) LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol Cell 48: 329–342
Mukherjee A, Patel B, Koga H, Cuervo AM, Jenny A (2016) Selective endosomal microautophagy is starvation‐inducible in Drosophila. Autophagy 12: 1984–1999
Munz C (2017) The macroautophagy machinery in endo‐ and exocytosis. J Mol Biol 429: 473–485
Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, Yasui H, Ueda H, Akazawa Y, Nakayama H, Taneike M, Misaka T, Omiya S, Shah AM, Yamamoto A, Nishida K, Ohsumi Y, Okamoto K, Sakata Y, Otsu K (2015) Bcl‐2‐like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun 6: 7527
Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen WL, Griffith J, Nag S, Wang K, Moss T, Baba M, McNew JA, Jiang X, Reggiori F, Melia TJ, Klionsky DJ (2011) SNARE proteins are required for macroautophagy. Cell 146: 290–302
Nakahira K, Pabon Porras MA, Choi AM (2016) Autophagy in pulmonary diseases. Am J Respir Crit Care Med 194: 1196–1207
Nakamoto M, Moy RH, Xu J, Bambina S, Yasunaga A, Shelly SS, Gold B, Cherry S (2012) Virus recognition by Toll‐7 activates antiviral autophagy in Drosophila. Immunity 36: 658–667
Nardacci R, Amendola A, Ciccosanti F, Corazzari M, Esposito V, Vlassi C, Taibi C, Fimia GM, Del Nonno F, Ippolito G, D'Offizi G, Piacentini M (2014) Autophagy plays an important role in the containment of HIV‐1 in nonprogressor‐infected patients. Autophagy 10: 1167–1178
Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ (2010) p62/SQSTM1 is required for Parkin‐induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6: 1090–1106
Nazarko TY, Ozeki K, Till A, Ramakrishnan G, Lotfi P, Yan M, Subramani S (2014) Peroxisomal Atg37 binds Atg30 or palmitoyl‐CoA to regulate phagophore formation during pexophagy. J Cell Biol 204: 541–557
Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, Cecconi F (2013) mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self‐association and function through AMBRA1 and TRAF6. Nat Cell Biol 15: 406–416
Neefjes J, Jongsma ML, Paul P, Bakke O (2011) Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 11: 823–836
Nguyen TN, Padman BS, Usher J, Oorschot V, Ramm G, Lazarou M (2016) Atg8 family LC3/GABARAP proteins are crucial for autophagosome‐lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol 215: 857–874
Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO (2009) Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136: 521–534
Niki T, Saito S, Gladish DK (2014) Granular bodies in root primary meristem cells of Zea mays L. var. Cuscoensis K. (Poaceae) that enter young vacuoles by invagination: a novel ribophagy mechanism. Protoplasma 251: 1141–1149
Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S (2009) Discovery of Atg5/Atg7‐independent alternative macroautophagy. Nature 461: 654–658
Niso‐Santano M, Malik SA, Pietrocola F, Bravo‐San Pedro JM, Marino G, Cianfanelli V, Ben‐Younes A, Troncoso R, Markaki M, Sica V, Izzo V, Chaba K, Bauvy C, Dupont N, Kepp O, Rockenfeller P, Wolinski H, Madeo F, Lavandero S, Codogno P et al (2015) Unsaturated fatty acids induce non‐canonical autophagy. EMBO J 34: 1025–1041
Noda NN, Inagaki F (2015) Mechanisms of autophagy. Annu Rev Biophys 44: 101–122
Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Lohr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dotsch V, Ney PA, Dikic I (2010) Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11: 45–51
Ogawa M, Yoshikawa Y, Kobayashi T, Mimuro H, Fukumatsu M, Kiga K, Piao Z, Ashida H, Yoshida M, Kakuta S, Koyama T, Goto Y, Nagatake T, Nagai S, Kiyono H, Kawalec M, Reichhart JM, Sasakawa C (2011) A Tecpr1‐dependent selective autophagy pathway targets bacterial pathogens. Cell Host Microbe 9: 376–389
Okamoto K, Kondo‐Okamoto N, Ohsumi Y (2009) Mitochondria‐anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 17: 87–97
Olsvik HL, Lamark T, Takagi K, Larsen KB, Evjen G, Overvatn A, Mizushima T, Johansen T (2015) FYCO1 contains a C‐terminally extended, LC3A/B‐preferring LC3‐interacting region (LIR) motif required for efficient maturation of autophagosomes during basal autophagy. J Biol Chem 290: 29361–29374
Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez‐Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM (2013) Interplay of LRRK2 with chaperone‐mediated autophagy. Nat Neurosci 16: 394–406
O'Rourke EJ, Ruvkun G (2013) MXL‐3 and HLH‐30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat Cell Biol 15: 668–676
Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, Tooze SA (2012) Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 23: 1860–1873
Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B (2007) HSV‐1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1: 23–35
Orvedahl A, MacPherson S, Sumpter R Jr, Talloczy Z, Zou Z, Levine B (2010) Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 7: 115–127
Orvedahl A, Sumpter R Jr, Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, Forst CV, Wrana JL, Zhang YE, Luby‐Phelps K, Xavier RJ, Xie Y, Levine B (2011) Image‐based genome‐wide siRNA screen identifies selective autophagy factors. Nature 480: 113–117
Ossareh‐Nazari B, Bonizec M, Cohen M, Dokudovskaya S, Delalande F, Schaeffer C, Van Dorsselaer A, Dargemont C (2010) Cdc48 and Ufd3, new partners of the ubiquitin protease Ubp3, are required for ribophagy. EMBO Rep 11: 548–554
Ossareh‐Nazari B, Nino CA, Bengtson MH, Lee JW, Joazeiro CA, Dargemont C (2014) Ubiquitylation by the Ltn1 E3 ligase protects 60S ribosomes from starvation‐induced selective autophagy. J Cell Biol 204: 909–917
Palikaras K, Tavernarakis N (2014) Mitochondrial homeostasis: the interplay between mitophagy and mitochondrial biogenesis. Exp Gerontol 56: 182–188
Palikaras K, Lionaki E, Tavernarakis N (2015) Coordination of mitophagy and mitochondrial biogenesis during ageing in Caenorhabditis elegans. Nature 521: 525–528
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282: 24131–24145
Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, Maiolica A, Hansmann I, Pfaffenwimmer T, Kijanska M, Stoffel I, Lee SS, Brezovich A, Lou JH, Turk BE, Aebersold R, Ammerer G, Peter M, Kraft C (2014) Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell 53: 471–483
Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441: 1157–1161
Park YE, Hayashi YK, Bonne G, Arimura T, Noguchi S, Nonaka I, Nishino I (2009) Autophagic degradation of nuclear components in mammalian cells. Autophagy 5: 795–804
Park C, Suh Y, Cuervo AM (2015) Regulated degradation of Chk1 by chaperone‐mediated autophagy in response to DNA damage. Nat Commun 6: 6823
Park JM, Jung CH, Seo M, Otto NM, Grunwald D, Kim KH, Moriarity B, Kim YM, Starker C, Nho RS, Voytas D, Kim DH (2016) The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 12: 547–564
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl‐2 antiapoptotic proteins inhibit Beclin 1‐dependent autophagy. Cell 122: 927–939
Paul P, Munz C (2016) Autophagy and mammalian viruses: roles in immune response, viral replication, and beyond. Adv Virus Res 95: 149–195
Poklepovic A, Gewirtz DA (2014) Outcome of early clinical trials of the combination of hydroxychloroquine with chemotherapy in cancer. Autophagy 10: 1478–1480
Politi Y, Gal L, Kalifa Y, Ravid L, Elazar Z, Arama E (2014) Paternal mitochondrial destruction after fertilization is mediated by a common endocytic and autophagic pathway in Drosophila. Dev Cell 29: 305–320
Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbe S, Clague MJ, Tooze SA (2010) Mammalian Atg18 (WIPI2) localizes to omegasome‐anchored phagophores and positively regulates LC3 lipidation. Autophagy 6: 506–522
Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, Deretic V (2015) Secretory autophagy. Curr Opin Cell Biol 35: 106–116
Proikas‐Cezanne T, Takacs Z, Donnes P, Kohlbacher O (2015) WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci 128: 207–217
Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC (2013) Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 154: 1285–1299
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B (2003) Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 112: 1809–1820
Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC (2010) Plasma membrane contributes to the formation of pre‐autophagosomal structures. Nat Cell Biol 12: 747–757
Record M, Carayon K, Poirot M, Silvente‐Poirot S (2014) Exosomes as new vesicular lipid transporters involved in cell‐cell communication and various pathophysiologies. Biochim Biophys Acta 1841: 108–120
Reggiori F, Monastyrska I, Shintani T, Klionsky DJ (2005) The actin cytoskeleton is required for selective types of autophagy, but not nonspecific autophagy, in the yeast Saccharomyces cerevisiae. Mol Biol Cell 16: 5843–5856
Rello‐Varona S, Lissa D, Shen S, Niso‐Santano M, Senovilla L, Marino G, Vitale I, Jemaa M, Harper F, Pierron G, Castedo M, Kroemer G (2012) Autophagic removal of micronuclei. Cell Cycle 11: 170–176
Roberts P, Moshitch‐Moshkovitz S, Kvam E, O'Toole E, Winey M, Goldfarb DS (2003) Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae. Mol Biol Cell 14: 129–141
Rockel JS, Kapoor M (2016) Autophagy: controlling cell fate in rheumatic diseases. Nat Rev Rheumatol 12: 517–531
Rockenfeller P, Koska M, Pietrocola F, Minois N, Knittelfelder O, Sica V, Franz J, Carmona‐Gutierrez D, Kroemer G, Madeo F (2015) Phosphatidylethanolamine positively regulates autophagy and longevity. Cell Death Differ 22: 499–508
Rodriguez‐Muela N, Koga H, Garcia‐Ledo L, de la Villa P, de la Rosa EJ, Cuervo AM, Boya P (2013) Balance between autophagic pathways preserves retinal homeostasis. Aging Cell 12: 478–488
Rogov V, Dotsch V, Johansen T, Kirkin V (2014) Interactions between autophagy receptors and ubiquitin‐like proteins form the molecular basis for selective autophagy. Mol Cell 53: 167–178
Rojansky R, Cha MY, Chan DC (2016) Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. Elife 5: e17896
Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, McAfee Q, Fisher J, Troxel AB, Piao S, Heitjan DF, Tan KS, Pontiggia L, O'Dwyer PJ, Davis LE, Amaravadi RK (2014) A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 10: 1359–1368
Rossin F, D'Eletto M, Falasca L, Sepe S, Cocco S, Fimia GM, Campanella M, Mastroberardino PG, Farrace MG, Piacentini M (2015) Transglutaminase 2 ablation leads to mitophagy impairment associated with a metabolic shift towards aerobic glycolysis. Cell Death Differ 22: 408–418
Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A (2011) The autophagy protein Atg12 associates with antiapoptotic Bcl‐2 family members to promote mitochondrial apoptosis. Mol Cell 44: 698–709
Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL (2013) ULK1 induces autophagy by phosphorylating Beclin‐1 and activating VPS34 lipid kinase. Nat Cell Biol 15: 741–750
Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, Potolicchio I, Nieves E, Cuervo AM, Santambrogio L (2011) Microautophagy of cytosolic proteins by late endosomes. Dev Cell 20: 131–139
Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin‐induced IL‐1beta production. Nature 456: 264–268
Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, Yamamoto N, Kawai T, Ishii K, Takeuchi O, Yoshimori T, Akira S (2009) Atg9a controls dsDNA‐driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA 106: 20842–20846
Saleh T, Cuttino L, Gewirtz DA (2016) Autophagy is not uniformly cytoprotective: a personalized medicine approach for autophagy inhibition as a therapeutic strategy in non‐small cell lung cancer. Biochim Biophys Acta 1860: 2130–2136
Salvador N, Aguado C, Horst M, Knecht E (2000) Import of a cytosolic protein into lysosomes by chaperone‐mediated autophagy depends on its folding state. J Biol Chem 275: 27447–27456
Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J (2008) Essential role for Nix in autophagic maturation of erythroid cells. Nature 454: 232–235
Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, Green DR (2007) Toll‐like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450: 1253–1257
Sargent G, van Zutphen T, Shatseva T, Zhang L, Di Giovanni V, Bandsma R, Kim PK (2016) PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J Cell Biol 214: 677–690
Sato M, Sato K (2011) Degradation of paternal mitochondria by fertilization‐triggered autophagy in Caenorhabditis elegans embryos. Science 334: 1141–1144
Schlegel A, Giddings TH Jr, Ladinsky MS, Kirkegaard K (1996) Cellular origin and ultrastructure of membranes induced during poliovirus infection. J Virol 70: 6576–6588
Schneider JL, Suh Y, Cuervo AM (2014) Deficient chaperone‐mediated autophagy in liver leads to metabolic dysregulation. Cell Metab 20: 417–432
Schneider JL, Villarroya J, Diaz‐Carretero A, Patel B, Urbanska AM, Thi MM, Villarroya F, Santambrogio L, Cuervo AM (2015) Loss of hepatic chaperone‐mediated autophagy accelerates proteostasis failure in aging. Aging Cell 14: 249–264
Schuck S, Gallagher CM, Walter P (2014) ER‐phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J Cell Sci 127: 4078–4088
Schweichel JU, Merker HJ (1973) The morphology of various types of cell death in prenatal tissues. Teratology 7: 253–266
Scott SV, Hefner‐Gravink A, Morano KA, Noda T, Ohsumi Y, Klionsky DJ (1996) Cytoplasm‐to‐vacuole targeting and autophagy employ the same machinery to deliver proteins to the yeast vacuole. Proc Natl Acad Sci USA 93: 12304–12308
Scott SV, Nice DC III, Nau JJ, Weisman LS, Kamada Y, Keizer‐Gunnink I, Funakoshi T, Veenhuis M, Ohsumi Y, Klionsky DJ (2000) Apg13p and Vac8p are part of a complex of phosphoproteins that are required for cytoplasm to vacuole targeting. J Biol Chem 275: 25840–25849
Scott SV, Guan J, Hutchins MU, Kim J, Klionsky DJ (2001) Cvt19 is a receptor for the cytoplasm‐to‐vacuole targeting pathway. Mol Cell 7: 1131–1141
Seay MD, Dinesh‐Kumar SP (2005) Life after death: are autophagy genes involved in cell death and survival during plant innate immune responses? Autophagy 1: 185–186
Selleck EM, Orchard RC, Lassen KG, Beatty WL, Xavier RJ, Levine B, Virgin HW, Sibley LD (2015) A noncanonical autophagy pathway restricts Toxoplasma gondii growth in a strain‐specific manner in IFN‐gamma‐activated human cells. MBio 6: e01157‐15
Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S, Sun X, Yoon G, Kang Y, Zhong W, Ma J, Kemper B, Kemper JK (2014) Transcriptional regulation of autophagy by an FXR‐CREB axis. Nature 516: 108–111
Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A (2011) TFEB links autophagy to lysosomal biogenesis. Science 332: 1429–1433
Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A (2012) A lysosome‐to‐nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31: 1095–1108
Settembre C, Fraldi A, Medina DL, Ballabio A (2013) Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 14: 283–296
Sharma K, Le N, Alotaibi M, Gewirtz DA (2014) Cytotoxic autophagy in cancer therapy. Int J Mol Sci 15: 10034–10051
Shcherbik N, Pestov DG (2011) The ubiquitin ligase Rsp5 is required for ribosome stability in Saccharomyces cerevisiae. RNA 17: 1422–1428
Shibutani ST, Yoshimori T (2014) A current perspective of autophagosome biogenesis. Cell Res 24: 58–68
Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J (2016) Aging and autophagy in the heart. Circ Res 118: 1563–1576
Shpilka T, Weidberg H, Pietrokovski S, Elazar Z (2011) Atg8: an autophagy‐related ubiquitin‐like protein family. Genome Biol 12: 226
Shravage BV, Hill JH, Powers CM, Wu L, Baehrecke EH (2013) Atg6 is required for multiple vesicle trafficking pathways and hematopoiesis in Drosophila. Development 140: 1321–1329
Shvets E, Elazar Z (2008) Autophagy‐independent incorporation of GFP‐LC3 into protein aggregates is dependent on its interaction with p62/SQSTM1. Autophagy 4: 1054–1056
Sica V, Galluzzi L, Bravo‐San Pedro JM, Izzo V, Maiuri MC, Kroemer G (2015) Organelle‐specific initiation of autophagy. Mol Cell 59: 522–539
Simonsen A, Birkeland HC, Gillooly DJ, Mizushima N, Kuma A, Yoshimori T, Slagsvold T, Brech A, Stenmark H (2004) Alfy, a novel FYVE‐domain‐containing protein associated with protein granules and autophagic membranes. J Cell Sci 117: 4239–4251
Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD (2008) Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4: 176–184
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ (2009) Autophagy regulates lipid metabolism. Nature 458: 1131–1135
Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, Sawada N, Yamada A, Mizushima N, Uchiyama Y, Kominami E, Tanaka K, Komatsu M (2008) The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell 19: 4762–4775
Spinnenhirn V, Farhan H, Basler M, Aichem A, Canaan A, Groettrup M (2014) The ubiquitin‐like modifier FAT10 decorates autophagy‐targeted Salmonella and contributes to Salmonella resistance in mice. J Cell Sci 127: 4883–4893
Sriram SM, Kim BY, Kwon YT (2011) The N‐end rule pathway: emerging functions and molecular principles of substrate recognition. Nat Rev Mol Cell Biol 12: 735–747
Stanley RE, Ragusa MJ, Hurley JH (2014) The beginning of the end: how scaffolds nucleate autophagosome biogenesis. Trends Cell Biol 24: 73–81
Stolz A, Ernst A, Dikic I (2014) Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16: 495–501
Sumpter R Jr, Sirasanagandla S, Fernandez AF, Wei Y, Dong X, Franco L, Zou Z, Marchal C, Lee MY, Clapp DW, Hanenberg H, Levine B (2016) Fanconi anemia proteins function in mitophagy and immunity. Cell 165: 867–881
Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q (2008) Identification of Barkor as a mammalian autophagy‐specific factor for Beclin 1 and class III phosphatidylinositol 3‐kinase. Proc Natl Acad Sci USA 105: 19211–19216
Talloczy Z, Jiang W, Virgin H, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B (2002) Regulation of starvation‐ and virus‐induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA 99: 190–195
Tanaka C, Tan LJ, Mochida K, Kirisako H, Koizumi M, Asai E, Sakoh‐Nakatogawa M, Ohsumi Y, Nakatogawa H (2014) Hrr25 triggers selective autophagy‐related pathways by phosphorylating receptor proteins. J Cell Biol 207: 91–105
Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q (2013) Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 502: 254–257
Tasset I, Cuervo AM (2016) Role of chaperone‐mediated autophagy in metabolism. FEBS J 283: 2403–2413
Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F (2009) The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin‐coated bacteria. Nat Immunol 10: 1215–1221
Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F (2012) Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482: 414–418
Thurston TL, Boyle KB, Allen M, Ravenhill BJ, Karpiyevich M, Bloor S, Kaul A, Noad J, Foeglein A, Matthews SA, Komander D, Bycroft M, Randow F (2016) Recruitment of TBK1 to cytosol‐invading Salmonella induces WIPI2‐dependent antibacterial autophagy. EMBO J 35: 1779–1792
Tian Y, Li Z, Hu W, Ren H, Tian E, Zhao Y, Lu Q, Huang X, Yang P, Li X, Wang X, Kovacs AL, Yu L, Zhang H (2010) Caenorhabditis elegans screen identifies autophagy genes specific to multicellular organisms. Cell 141: 1042–1055
Tooze SA, Abada A, Elazar Z (2014) Endocytosis and autophagy: exploitation or cooperation? Cold Spring Harb Perspect Biol 6: a018358
Torggler R, Papinski D, Brach T, Bas L, Schuschnig M, Pfaffenwimmer T, Rohringer S, Matzhold T, Schweida D, Brezovich A, Kraft C (2016) Two independent pathways within selective autophagy converge to activate Atg1 kinase at the vacuole. Mol Cell 64: 221–235
Toyooka K, Moriyasu Y, Goto Y, Takeuchi M, Fukuda H, Matsuoka K (2006) Protein aggregates are transported to vacuoles by a macroautophagic mechanism in nutrient‐starved plant cells. Autophagy 2: 96–106
Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, Boneca IG, Allaoui A, Jones NL, Nunez G, Girardin SE, Philpott DJ (2010) Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 11: 55–62
Tsuboyama K, Koyama‐Honda I, Sakamaki Y, Koike M, Morishita H, Mizushima N (2016) The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 354: 1036–1041
Tsukada M, Ohsumi Y (1993) Isolation and characterization of autophagy‐defective mutants of Saccharomyces cerevisiae. FEBS Lett 333: 169–174
Tumbarello DA, Manna PT, Allen M, Bycroft M, Arden SD, Kendrick‐Jones J, Buss F (2015) The autophagy receptor TAX1BP1 and the molecular motor myosin VI are required for clearance of Salmonella Typhimurium by autophagy. PLoS Pathog 11: e1005174
Umekawa M, Klionsky DJ (2012) The cytoplasm‐to‐vacuole targeting pathway: a historical perspective. Int J Cell Biol 2012: 142634
Uttenweiler A, Mayer A (2008) Microautophagy in the yeast Saccharomyces cerevisiae. Methods Mol Biol 445: 245–259
Uytterhoeven V, Lauwers E, Maes I, Miskiewicz K, Melo MN, Swerts J, Kuenen S, Wittocx R, Corthout N, Marrink SJ, Munck S, Verstreken P (2015) Hsc70‐4 deforms membranes to promote synaptic protein turnover by endosomal microautophagy. Neuron 88: 735–748
Vakifahmetoglu‐Norberg H, Xia HG, Yuan J (2015) Pharmacologic agents targeting autophagy. J Clin Invest 125: 5–13
Valdor R, Mocholi E, Botbol Y, Guerrero‐Ros I, Chandra D, Koga H, Gravekamp C, Cuervo AM, Macian F (2014) Chaperone‐mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat Immunol 15: 1046–1054
Verlhac P, Gregoire IP, Azocar O, Petkova DS, Baguet J, Viret C, Faure M (2015) Autophagy receptor NDP52 regulates pathogen‐containing autophagosome maturation. Cell Host Microbe 17: 515–525
Vevea JD, Garcia EJ, Chan RB, Zhou B, Schultz M, Di Paolo G, McCaffery JM, Pon LA (2015) Role for lipid droplet biogenesis and microlipophagy in adaptation to lipid imbalance in yeast. Dev Cell 35: 584–599
Vicinanza M, Korolchuk VI, Ashkenazi A, Puri C, Menzies FM, Clarke JH, Rubinsztein DC (2015) PI(5)P regulates autophagosome biogenesis. Mol Cell 57: 219–234
Waite KA, De‐La Mota‐Peynado A, Vontz G, Roelofs J (2016) Starvation induces proteasome autophagy with different pathways for core and regulatory particles. J Biol Chem 291: 3239–3253
Waliullah TM, Yeasmin AM, Kaneko A, Koike N, Terasawa M, Totsuka T, Ushimaru T (2017) Rim15 and Sch9 kinases are involved in induction of autophagic degradation of ribosomes in budding yeast. Biosci Biotechnol Biochem 81: 307–310
Walter KM, Schonenberger MJ, Trotzmuller M, Horn M, Elsasser HP, Moser AB, Lucas MS, Schwarz T, Gerber PA, Faust PL, Moch H, Kofeler HC, Krek W, Kovacs WJ (2014) Hif‐2alpha promotes degradation of mammalian peroxisomes by selective autophagy. Cell Metab 20: 882–897
Wang CW, Miao YH, Chang YS (2014) A sterol‐enriched vacuolar microdomain mediates stationary phase lipophagy in budding yeast. J Cell Biol 206: 357–366
Wang Y, Zhang Y, Chen L, Liang Q, Yin XM, Miao L, Kang BH, Xue D (2016) Kinetics and specificity of paternal mitochondrial elimination in Caenorhabditis elegans. Nat Commun 7: 12569
Weckman A, Di Ieva A, Rotondo F, Syro LV, Ortiz LD, Kovacs K, Cusimano MD (2014) Autophagy in the endocrine glands. J Mol Endocrinol 52: R151–R163
Wei Y, Chiang WC, Sumpter R Jr, Mishra P, Levine B (2017) Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168: 224–238
Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dotsch V, Bumann D, Dikic I (2011) Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333: 228–233
Wild P, McEwan DG, Dikic I (2014) The LC3 interactome at a glance. J Cell Sci 127: 3–9
Wilkinson DS, Jariwala JS, Anderson E, Mitra K, Meisenhelder J, Chang JT, Ideker T, Hunter T, Nizet V, Dillin A, Hansen M (2015) Phosphorylation of LC3 by the Hippo kinases STK3/STK4 is essential for autophagy. Mol Cell 57: 55–68
Wing SS, Chiang HL, Goldberg AL, Dice JF (1991) Proteins containing peptide sequences related to Lys‐Phe‐Glu‐Arg‐Gln are selectively depleted in liver and heart, but not skeletal muscle, of fasted rats. Biochem J 275(Pt 1): 165–169
Wold MS, Lim J, Lachance V, Deng Z, Yue Z (2016) ULK1‐mediated phosphorylation of ATG14 promotes autophagy and is impaired in Huntington's disease models. Mol Neurodegener 11: 76
Wong YC, Holzbaur EL (2014) Optineurin is an autophagy receptor for damaged mitochondria in parkin‐mediated mitophagy that is disrupted by an ALS‐linked mutation. Proc Natl Acad Sci USA 111: E4439–E4448
Wu J, Randle KE, Wu LP (2007) ird1 is a Vps15 homologue important for antibacterial immune responses in Drosophila. Cell Microbiol 9: 1073–1085
Xie C, Ginet V, Sun Y, Koike M, Zhou K, Li T, Li H, Li Q, Wang X, Uchiyama Y, Truttmann AC, Kroemer G, Puyal J, Blomgren K, Zhu C (2016) Neuroprotection by selective neuronal deletion of Atg7 in neonatal brain injury. Autophagy 12: 410–423
Xu T, Nicolson S, Denton D, Kumar S (2015) Distinct requirements of Autophagy‐related genes in programmed cell death. Cell Death Differ 22: 1792–1802
Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo‐Kakuta C, Ichikawa R, Kinjo M, Ohsumi Y (2012) Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol 198: 219–233
Yamashita S, Abe K, Tatemichi Y, Fujiki Y (2014) The membrane peroxin PEX3 induces peroxisome‐ubiquitination‐linked pexophagy. Autophagy 10: 1549–1564
Yang Z, Klionsky DJ (2010) Eaten alive: a history of macroautophagy. Nat Cell Biol 12: 814–822
Yano T, Mita S, Ohmori H, Oshima Y, Fujimoto Y, Ueda R, Takada H, Goldman WE, Fukase K, Silverman N, Yoshimori T, Kurata S (2008) Autophagic control of listeria through intracellular innate immune recognition in Drosophila. Nat Immunol 9: 908–916
Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU (2006) Calpain‐mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 8: 1124–1132
Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, Hailey DW, Oorschot V, Klumperman J, Baehrecke EH, Lenardo MJ (2010) Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465: 942–946
Yue Z, Jin S, Yang C, Levine AJ, Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 100: 15077–15082
Zaffagnini G, Martens S (2016) Mechanisms of selective autophagy. J Mol Biol 428: 1714–1724
Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas‐Kramarski R, Kimchi A (2009) DAP‐kinase‐mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl‐XL and induction of autophagy. EMBO Rep 10: 285–292
Zhang J, Tripathi DN, Jing J, Alexander A, Kim J, Powell RT, Dere R, Tait‐Mulder J, Lee JH, Paull TT, Pandita RK, Charaka VK, Pandita TK, Kastan MB, Walker CL (2015) ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat Cell Biol 17: 1259–1269
Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, Cadwell K, Delgado MA, Ponpuak M, Green KG, Schmidt RE, Mizushima N, Deretic V, Sibley LD, Virgin HW (2008) Autophagosome‐independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe 4: 458–469
Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z (2009) Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1‐phosphatidylinositol‐3‐kinase complex. Nat Cell Biol 11: 468–476
Zhong Z, Sanchez‐Lopez E, Karin M (2016) Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell 166: 288–298
Zhou Q, Li H, Li H, Nakagawa A, Lin JL, Lee ES, Harry BL, Skeen‐Gaar RR, Suehiro Y, William D, Mitani S, Yuan HS, Kang BH, Xue D (2016) Mitochondrial endonuclease G mediates breakdown of paternal mitochondria upon fertilization. Science 353: 394–399
Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT (2007) Regulation of autophagy by extracellular signal‐regulated protein kinases during 1‐methyl‐4‐phenylpyridinium‐induced cell death. Am J Pathol 170: 75–86
Zhu J, Wang KZ, Chu CT (2013) After the banquet: mitochondrial biogenesis, mitophagy, and cell survival. Autophagy 9: 1663–1676
Zou CG, Ma YC, Dai LL, Zhang KQ (2014) Autophagy protects Caenorhabditis elegans against necrosis during Pseudomonas aeruginosa infection. Proc Natl Acad Sci USA 111: 12480–12485
Zutphen T, Veenhuis M, van der Klei IJ (2008) Pex14 is the sole component of the peroxisomal translocon that is required for pexophagy. Autophagy 4: 63–66
Acknowledgements
We apologize to the authors of several high‐quality scientific articles that contributed significantly to the development of the field but could not be cited.
Author information
Authors and Affiliations
Contributions
LG conceived and wrote the manuscript, centralized and integrated comments from co‐authors, and revised the review upon editorial feedback. JMBSP designed the figure, performed bibliographic searches, and helped with table preparation. All authors corrected the article and provided valuable input to obtain a unified view. With the exception of LG and GK, authors are listed alphabetically, which does not reflect their relative contribution to the preparation of this article.
Corresponding author
Ethics declarations
A.C.K. is an inventor on patents pertaining to Kras regulated metabolic pathways, redox control pathways in pancreatic cancer, targeting GOT1 as a therapeutic approach, and the autophagic control of iron metabolism. A.C.K. is on the SAB of Cornerstone Pharmaceuticals and is a founder of Vescor Therapeutics. The other authors declare that they have no conflict of interest.
Additional information
The EMBO Journal (2017) 36: 1811–1836
Rights and permissions
Copyright: The Authors
About this article
Cite this article
Galluzzi, L., Baehrecke, E.H., Ballabio, A. et al. Molecular definitions of autophagy and related processes. EMBO J 36, 1811–1836 (2017). https://doi.org/10.15252/embj.201796697
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.15252/embj.201796697
